Multiomics Analysis: CBMC Cite-Seq (Validation on Donor 2)
Reference: https://satijalab.org/seurat/v3.1/multimodal_vignette.html
[1]:
import scanpy as sc
import os
import pandas as pd
import numpy as np
import pickle as pkl
import matplotlib as mpl
import matplotlib.pyplot as plt
import scipy.stats
sc.settings.verbosity = 3
Process Protein
[2]:
adt_adata = sc.AnnData(sc.read_mtx("../../CITE-seq/bmcn-adt.mtx").T)
adt_adata.obs = pd.read_csv("../../CITE-seq/bmnc-meta.csv")
adt_adata.var_names = pd.read_table("../../CITE-seq/bmcn-adt-features.txt", index_col=0, header=None).index.tolist()
[3]:
adt_adata
[3]:
AnnData object with n_obs × n_vars = 30672 × 25
obs: 'Unnamed: 0', 'orig.ident', 'nCount_RNA', 'nFeature_RNA', 'nCount_ADT', 'nFeature_ADT', 'lane', 'donor', 'celltype.l1', 'celltype.l2', 'RNA.weight'
[4]:
adt_adata.obs.head()
[4]:
| Unnamed: 0 | orig.ident | nCount_RNA | nFeature_RNA | nCount_ADT | nFeature_ADT | lane | donor | celltype.l1 | celltype.l2 | RNA.weight | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | a_AAACCTGAGCTTATCG-1 | bmcite | 7546 | 2136 | 1350 | 25 | HumanHTO4 | batch1 | Progenitor cells | Prog_RBC | 0.482701 |
| 1 | a_AAACCTGAGGTGGGTT-1 | bmcite | 1029 | 437 | 2970 | 25 | HumanHTO1 | batch1 | T cell | gdT | 0.241789 |
| 2 | a_AAACCTGAGTACATGA-1 | bmcite | 1111 | 429 | 2474 | 23 | HumanHTO5 | batch1 | T cell | CD4 Naive | 0.507714 |
| 3 | a_AAACCTGCAAACCTAC-1 | bmcite | 2741 | 851 | 4799 | 25 | HumanHTO3 | batch1 | T cell | CD4 Memory | 0.431308 |
| 4 | a_AAACCTGCAAGGTGTG-1 | bmcite | 2099 | 843 | 5434 | 25 | HumanHTO2 | batch1 | Mono/DC | CD14 Mono | 0.568508 |
[5]:
ct = pd.crosstab(adt_adata.obs['celltype.l1'], adt_adata.obs['celltype.l2'])
ct.style
[5]:
| celltype.l2 | CD14 Mono | CD16 Mono | CD4 Memory | CD4 Naive | CD56 bright NK | CD8 Effector_1 | CD8 Effector_2 | CD8 Memory_1 | CD8 Memory_2 | CD8 Naive | GMP | HSC | LMPP | MAIT | Memory B | NK | Naive B | Plasmablast | Prog_B 1 | Prog_B 2 | Prog_DC | Prog_Mk | Prog_RBC | Treg | cDC2 | gdT | pDC |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| celltype.l1 | |||||||||||||||||||||||||||
| B cell | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1630 | 0 | 1900 | 223 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Mono/DC | 6486 | 433 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 482 | 0 | 328 |
| NK | 0 | 0 | 0 | 0 | 143 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1267 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Progenitor cells | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 748 | 263 | 292 | 0 | 0 | 0 | 0 | 0 | 145 | 129 | 263 | 124 | 915 | 0 | 0 | 0 | 0 |
| T cell | 0 | 0 | 3360 | 4500 | 0 | 577 | 307 | 444 | 555 | 3974 | 0 | 0 | 0 | 520 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 297 | 0 | 367 | 0 |
[6]:
groups = {i: ct.columns[ct.loc[i] > 0].tolist() for i in ct.index}
groups
[6]:
{'B cell': ['Memory B', 'Naive B', 'Plasmablast'],
'Mono/DC': ['CD14 Mono', 'CD16 Mono', 'cDC2', 'pDC'],
'NK': ['CD56 bright NK', 'NK'],
'Progenitor cells': ['GMP',
'HSC',
'LMPP',
'Prog_B 1',
'Prog_B 2',
'Prog_DC',
'Prog_Mk',
'Prog_RBC'],
'T cell': ['CD4 Memory',
'CD4 Naive',
'CD8 Effector_1',
'CD8 Effector_2',
'CD8 Memory_1',
'CD8 Memory_2',
'CD8 Naive',
'MAIT',
'Treg',
'gdT']}
[7]:
def f(l2):
if 'CD8 Effector' in l2:
return 'CD8 Effector'
elif 'CD8 Memory' in l2:
return 'CD8 Memory'
elif 'Prog_B' in l2:
return 'Prog_B'
else:
return l2
adt_adata.obs['celltype.l1.5'] = [f(i) for i in adt_adata.obs['celltype.l2']]
def f(l1, l1_5, t):
if l1 == t:
return l1_5
else:
return l1
for g in groups:
adt_adata.obs[g] = [f(*i, g) for i in zip(adt_adata.obs['celltype.l1'], adt_adata.obs['celltype.l1.5'])]
[8]:
def f(l1, l1_5):
if 'T cell' in l1:
return 'T ' + l1_5
elif 'Progenitor cells' in l1:
return l1_5
else:
return 'zzz'
adt_adata.obs['celltype.oi'] = [f(*i) for i in zip(adt_adata.obs['celltype.l1'], adt_adata.obs['celltype.l1.5'])]
[9]:
celltype_oi = sorted(list(set(adt_adata.obs['celltype.oi'].unique().tolist()) - {'zzz'}))
celltype_oi
[9]:
['GMP',
'HSC',
'LMPP',
'Prog_B',
'Prog_DC',
'Prog_Mk',
'Prog_RBC',
'T CD4 Memory',
'T CD4 Naive',
'T CD8 Effector',
'T CD8 Memory',
'T CD8 Naive',
'T MAIT',
'T Treg',
'T gdT']
[10]:
pd.crosstab(adt_adata.obs.donor, adt_adata.obs.lane)
[10]:
| lane | HumanHTO1 | HumanHTO10 | HumanHTO2 | HumanHTO3 | HumanHTO4 | HumanHTO5 | HumanHTO6 | HumanHTO7 | HumanHTO8 | HumanHTO9 |
|---|---|---|---|---|---|---|---|---|---|---|
| donor | ||||||||||
| batch1 | 1353 | 1317 | 1443 | 1437 | 1168 | 1427 | 1500 | 1766 | 1569 | 1488 |
| batch2 | 1512 | 1400 | 1643 | 1728 | 1318 | 1582 | 1662 | 1961 | 1803 | 1595 |
[11]:
adt_adata.obs.donor.value_counts()
[11]:
batch2 16204
batch1 14468
Name: donor, dtype: int64
[12]:
adt_adata.obs.lane.value_counts()
[12]:
HumanHTO7 3727
HumanHTO8 3372
HumanHTO3 3165
HumanHTO6 3162
HumanHTO2 3086
HumanHTO9 3083
HumanHTO5 3009
HumanHTO1 2865
HumanHTO10 2717
HumanHTO4 2486
Name: lane, dtype: int64
[13]:
adt_adata = adt_adata[adt_adata.obs.donor == 'batch2']
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1094: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if not is_categorical(df_full[k]):
[14]:
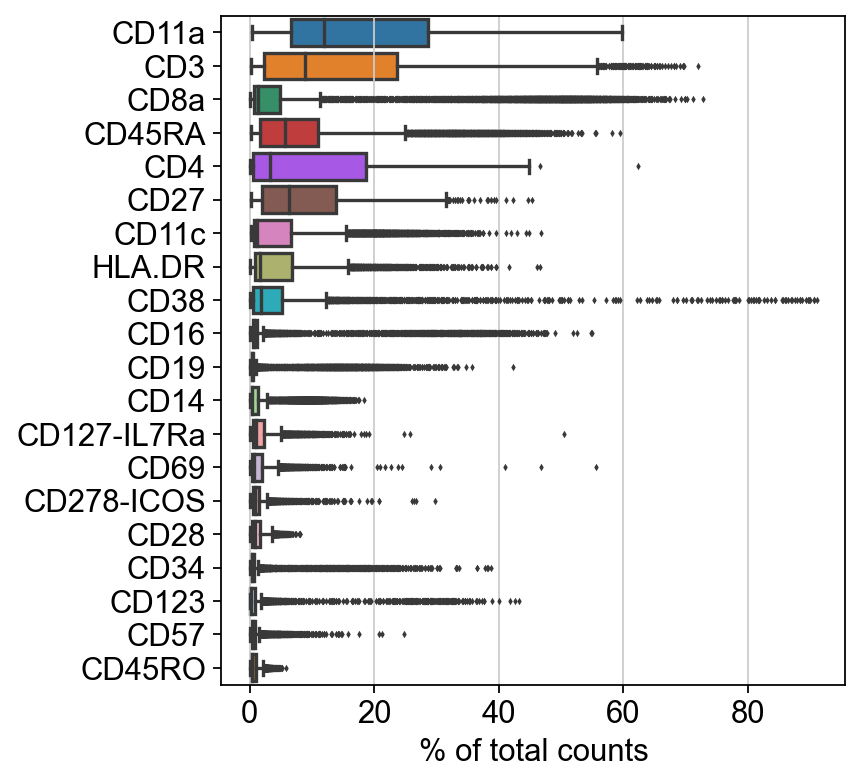
sc.settings.set_figure_params(dpi=80, facecolor='white')
sc.pl.highest_expr_genes(adt_adata, n_top=20)
normalizing counts per cell
finished ({time_passed})
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\scanpy\preprocessing\_normalization.py:138: UserWarning: Revieved a view of an AnnData. Making a copy.
view_to_actual(adata)
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:119: ImplicitModificationWarning: Transforming to str index.
warnings.warn("Transforming to str index.", ImplicitModificationWarning)
[15]:
sc.pp.normalize_total(adt_adata, target_sum=1e4)
sc.pp.log1p(adt_adata)
import scipy.stats.mstats
def clr(adata):
temp = np.array(adata.X.todense() + 1)
adata.X = np.log(temp / scipy.stats.mstats.gmean(temp, axis=1).reshape([-1, 1]))
# clr(adt_adata)
normalizing counts per cell
finished (0:00:00)
[16]:
sc.pp.scale(adt_adata, max_value=10)
... as `zero_center=True`, sparse input is densified and may lead to large memory consumption
[17]:
sc.tl.pca(adt_adata, svd_solver='arpack')
computing PCA
with n_comps=24
finished (0:00:00)
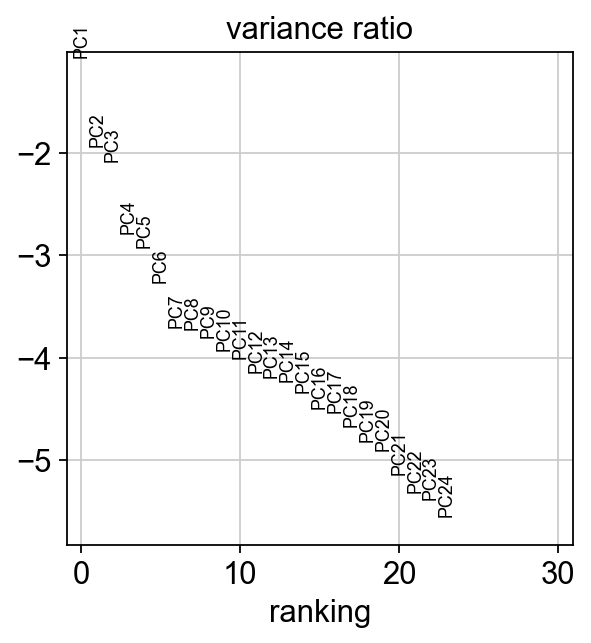
[18]:
sc.pl.pca_variance_ratio(adt_adata, log=True)
[19]:
sc.pp.neighbors(adt_adata, n_neighbors=50)
sc.tl.umap(adt_adata)
computing neighbors
using data matrix X directly
finished: added to `.uns['neighbors']`
`.obsp['distances']`, distances for each pair of neighbors
`.obsp['connectivities']`, weighted adjacency matrix (0:00:15)
computing UMAP
finished: added
'X_umap', UMAP coordinates (adata.obsm) (0:00:14)
[20]:
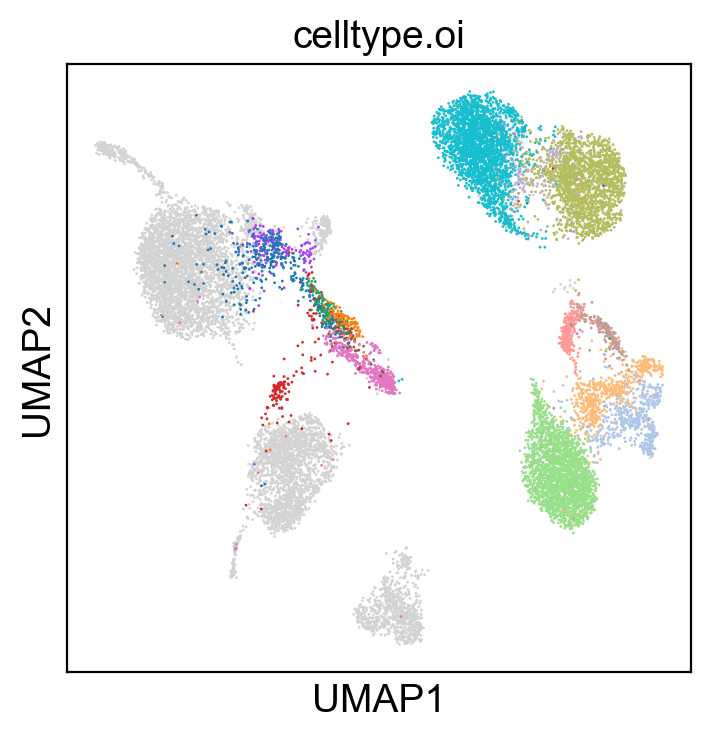
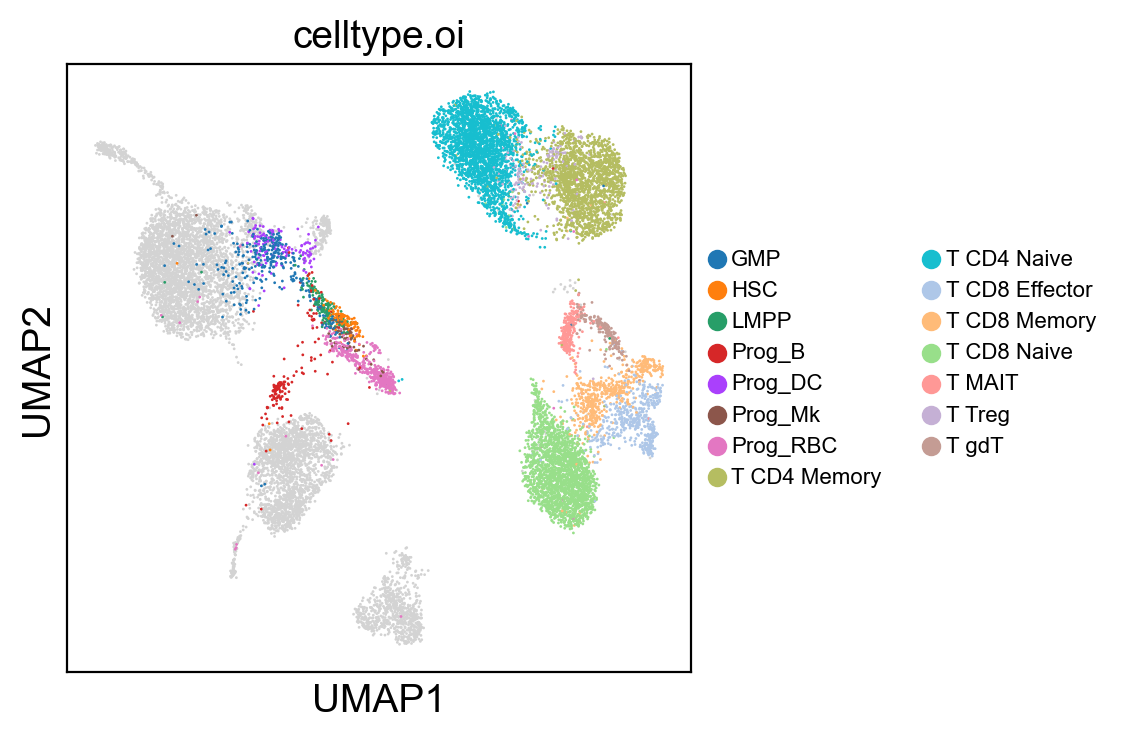
sc.settings.set_figure_params(dpi=100, facecolor='white')
sc.pl.umap(adt_adata, color=['celltype.oi'], groups=celltype_oi, legend_loc=None, legend_fontsize=8., size=4.)
sc.pl.umap(adt_adata, color=['celltype.oi'], groups=celltype_oi, legend_fontsize=8., size=4.)
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1192: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if is_string_dtype(df[key]) and not is_categorical(df[key])
... storing 'orig.ident' as categorical
... storing 'lane' as categorical
... storing 'donor' as categorical
... storing 'celltype.l1' as categorical
... storing 'celltype.l2' as categorical
... storing 'celltype.l1.5' as categorical
... storing 'B cell' as categorical
... storing 'Mono/DC' as categorical
... storing 'NK' as categorical
... storing 'Progenitor cells' as categorical
... storing 'T cell' as categorical
... storing 'celltype.oi' as categorical
[21]:
sc.settings.set_figure_params(dpi=100, facecolor='white')
sc.pl.umap(adt_adata, color=['celltype.l1.5'], legend_loc=None, legend_fontsize=8., size=4.)
sc.pl.umap(adt_adata, color=['celltype.l1.5'], legend_loc="on data", legend_fontsize=8., size=4.)
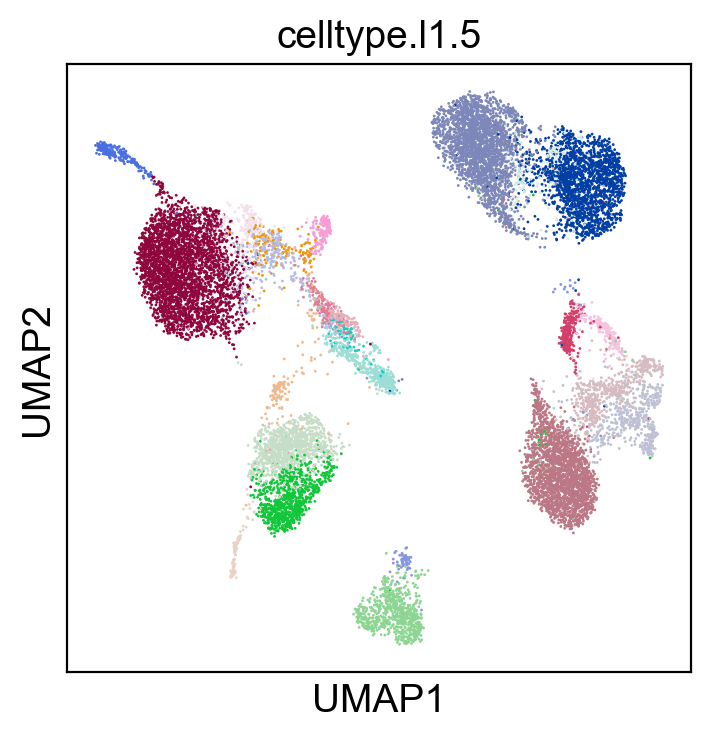
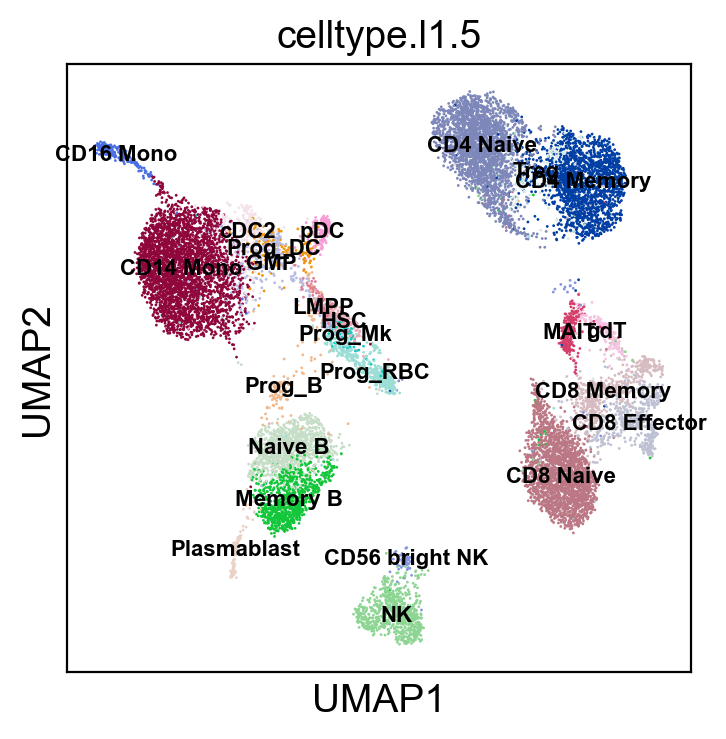
[22]:
sc.pl.umap(adt_adata, color=['T cell'], legend_loc=None, legend_fontsize=8., size=4.)
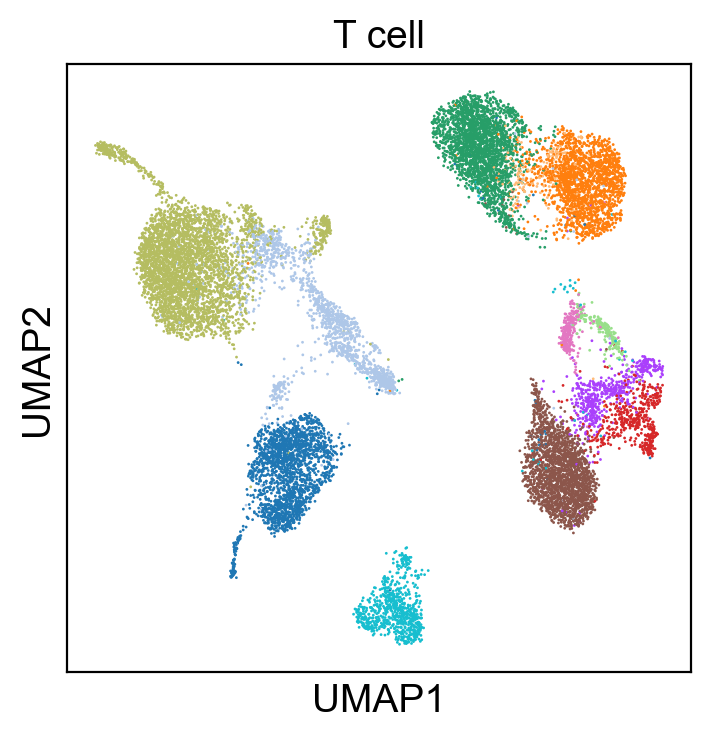
Process RNA
[23]:
rna_adata = sc.AnnData(sc.read_mtx("../../CITE-seq/bmcn-rna.mtx").T)
rna_adata.obs = pd.read_csv("../../CITE-seq/bmnc-meta.csv")
rna_adata.var_names = pd.read_table("../../CITE-seq/bmcn-rna-features.txt", index_col=0, header=None).index.tolist()
[24]:
rna_adata = rna_adata[rna_adata.obs.donor == 'batch2']
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1094: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if not is_categorical(df_full[k]):
[25]:
rna_adata.obs = adt_adata.obs.copy()
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:119: ImplicitModificationWarning: Transforming to str index.
warnings.warn("Transforming to str index.", ImplicitModificationWarning)
[26]:
sc.settings.set_figure_params(dpi=80, facecolor='white')
sc.pl.highest_expr_genes(rna_adata, n_top=20)
normalizing counts per cell
finished (0:00:00)
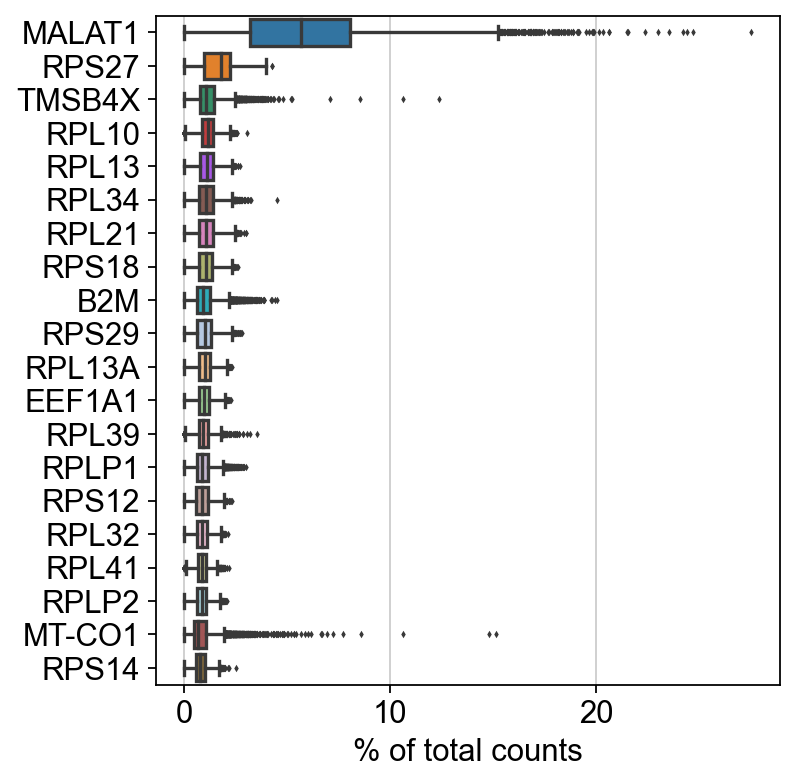
[27]:
sc.pp.normalize_total(rna_adata, target_sum=1e4)
sc.pp.log1p(rna_adata)
normalizing counts per cell
finished (0:00:00)
[28]:
#sc.pp.highly_variable_genes(rna_adata)
#sc.pl.highly_variable_genes(rna_adata)
#rna_adata.var.highly_variable.sum()
[29]:
#rna_adata = rna_adata[:, rna_adata.var.highly_variable]
[30]:
sc.pp.scale(rna_adata, max_value=10)
sc.tl.pca(rna_adata, svd_solver='arpack')
... as `zero_center=True`, sparse input is densified and may lead to large memory consumption
computing PCA
with n_comps=50
finished (0:00:25)
[31]:
sc.pl.pca_variance_ratio(rna_adata, log=True, n_pcs=50)
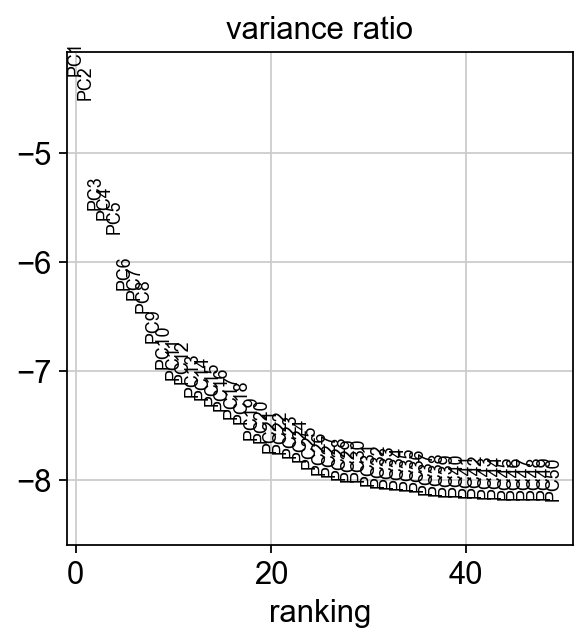
[32]:
sc.pp.neighbors(rna_adata, n_neighbors=40, n_pcs=50)
sc.tl.umap(rna_adata)
computing neighbors
using 'X_pca' with n_pcs = 50
finished: added to `.uns['neighbors']`
`.obsp['distances']`, distances for each pair of neighbors
`.obsp['connectivities']`, weighted adjacency matrix (0:00:11)
computing UMAP
finished: added
'X_umap', UMAP coordinates (adata.obsm) (0:00:13)
[33]:
sc.pl.umap(rna_adata, color=['celltype.oi'], groups=celltype_oi, legend_loc=None, legend_fontsize=8., size=4.)
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1192: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if is_string_dtype(df[key]) and not is_categorical(df[key])
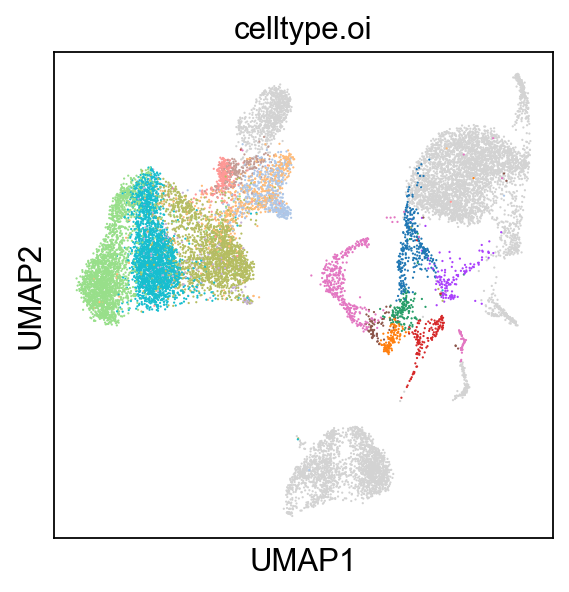
[34]:
sc.settings.set_figure_params(dpi=100, facecolor='white')
sc.pl.umap(rna_adata, color=['celltype.l1.5'], legend_loc="on data", legend_fontsize=8., size=4.)
sc.pl.umap(rna_adata, color=['celltype.l1.5'], legend_loc=None, legend_fontsize=8., size=4.)
sc.pl.umap(rna_adata, color=['celltype.l1.5'], legend_fontsize=8., size=4.)
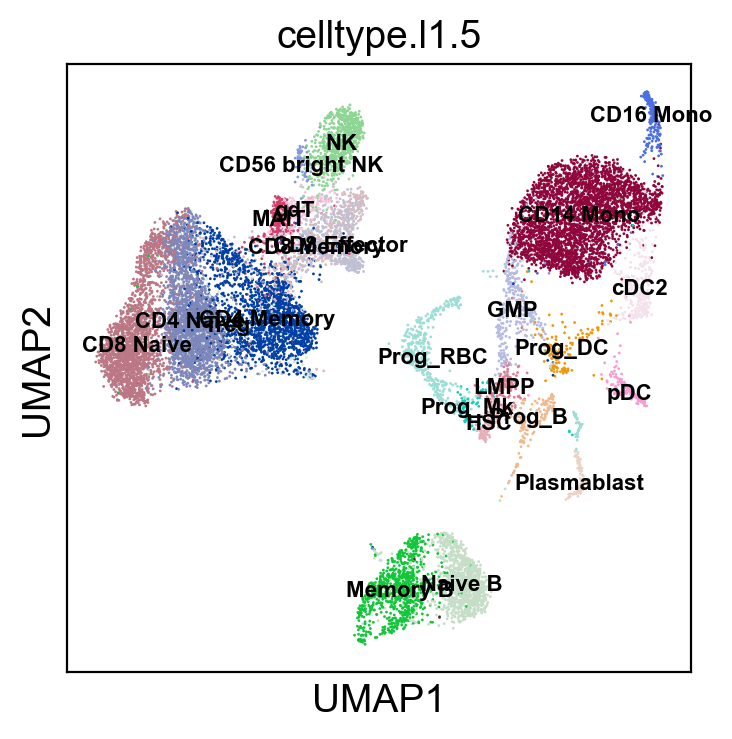
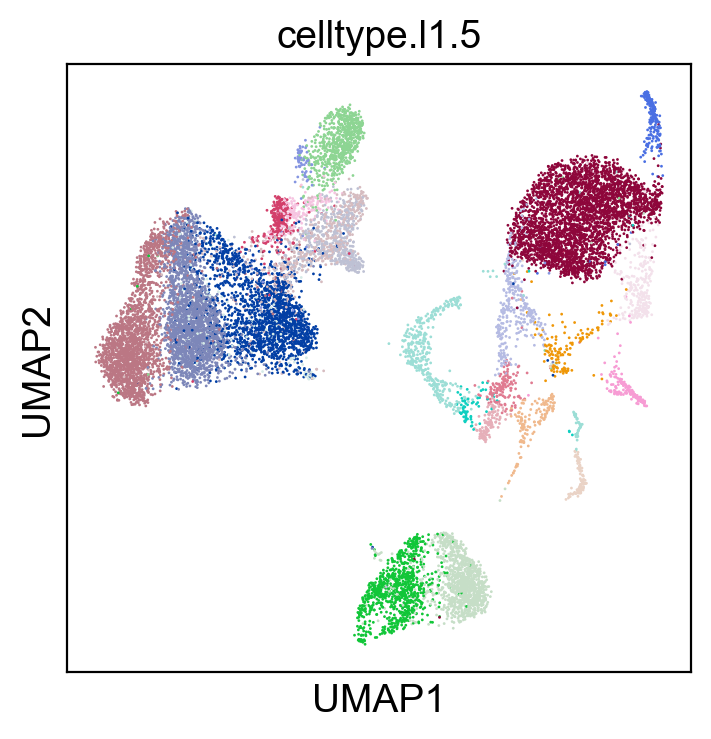
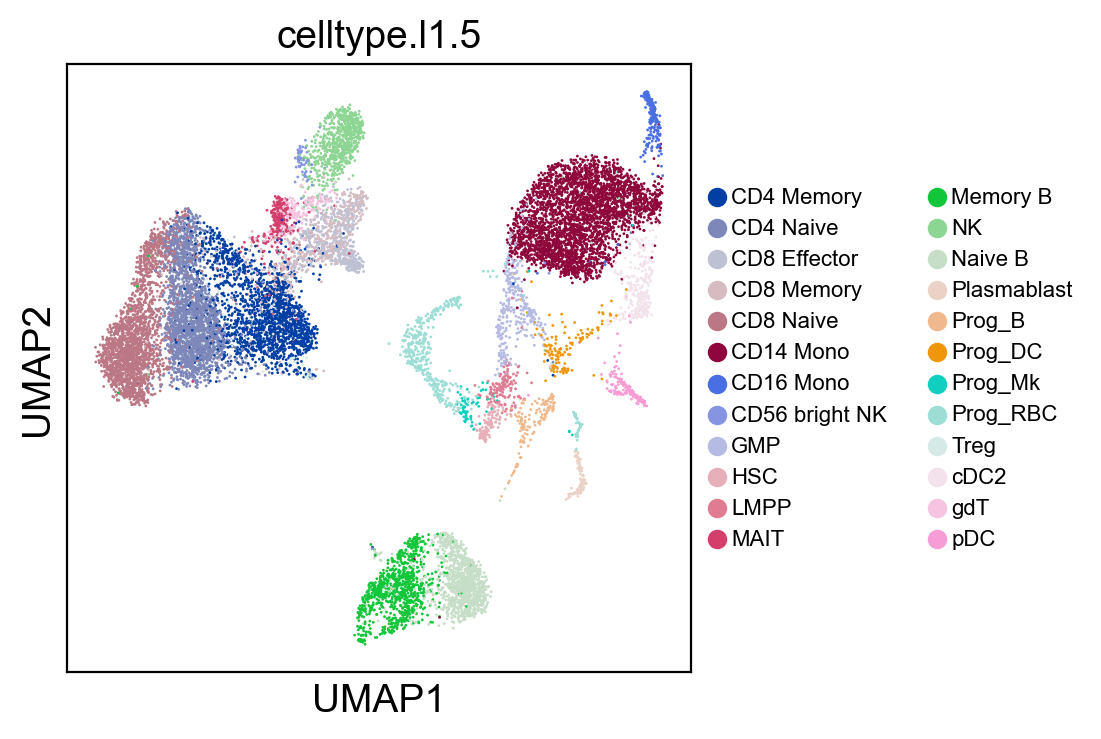
[35]:
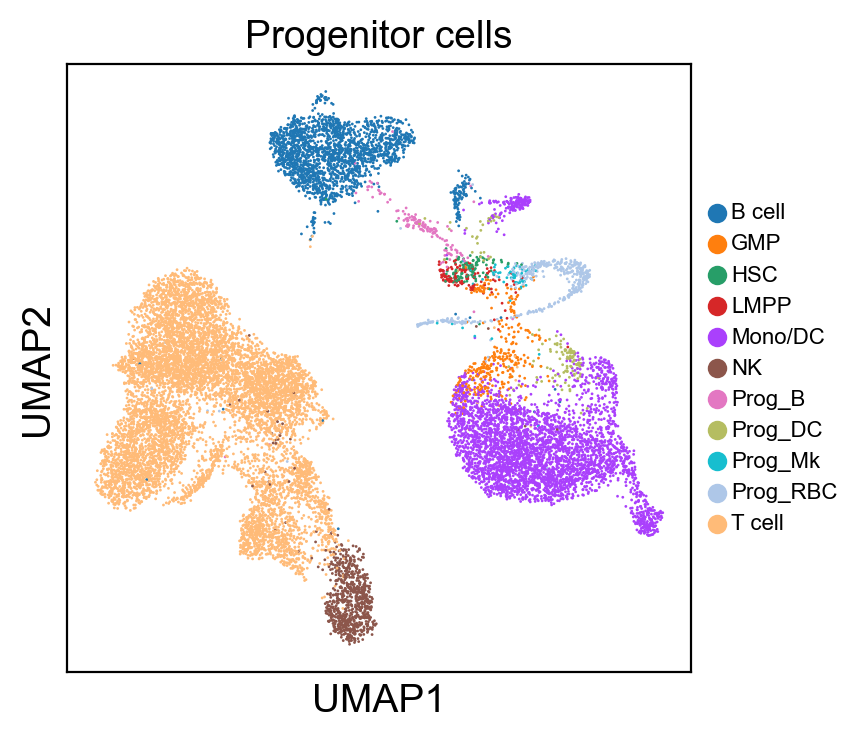
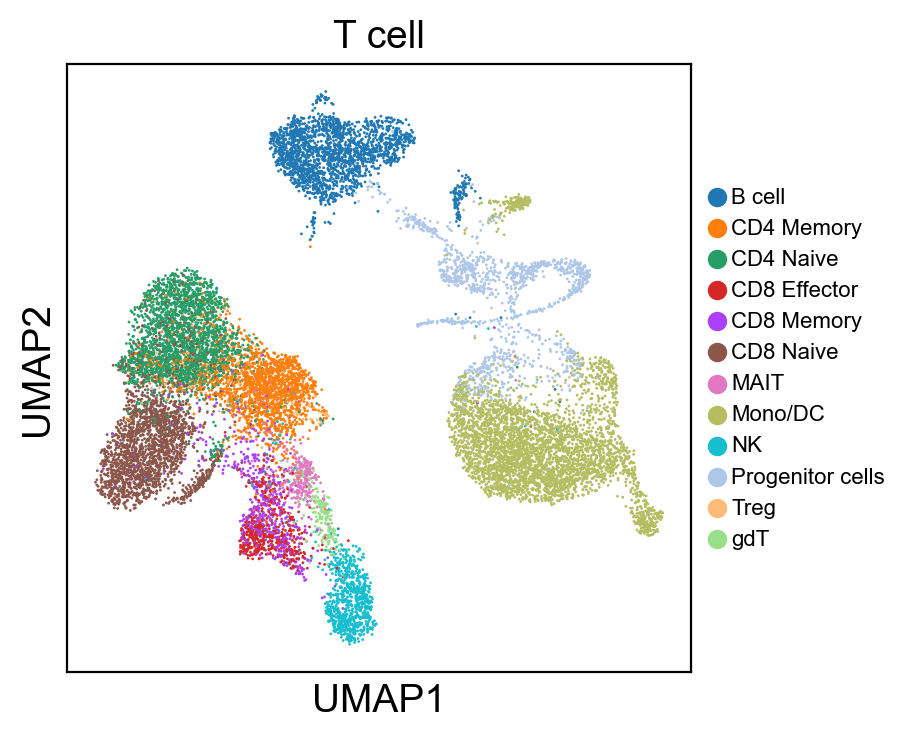
for g in groups:
sc.pl.umap(rna_adata, color=[g], legend_fontsize=8., size=4.)
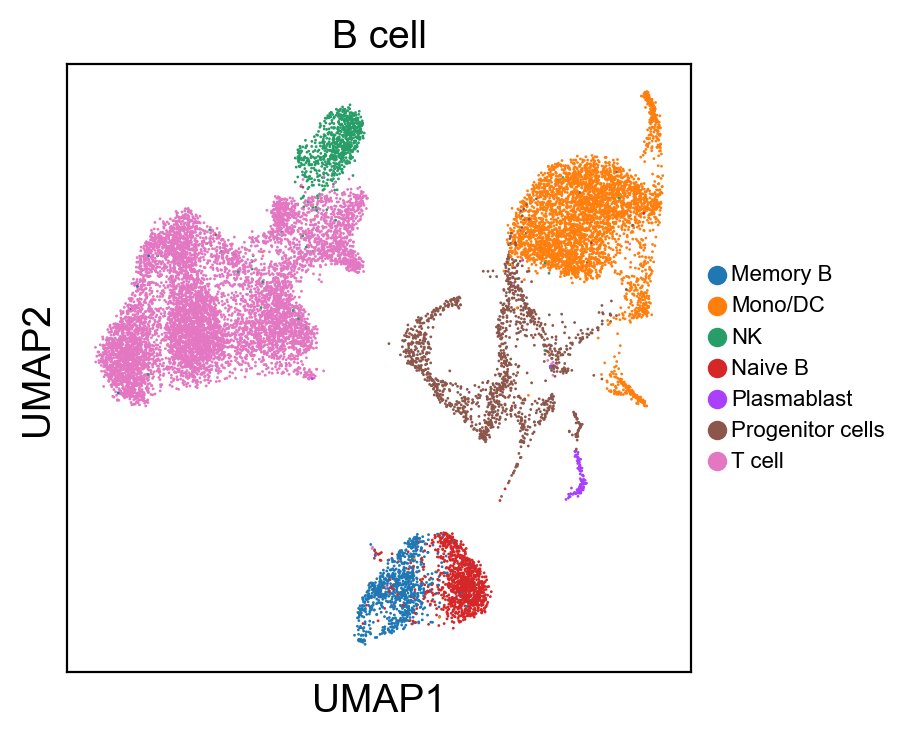
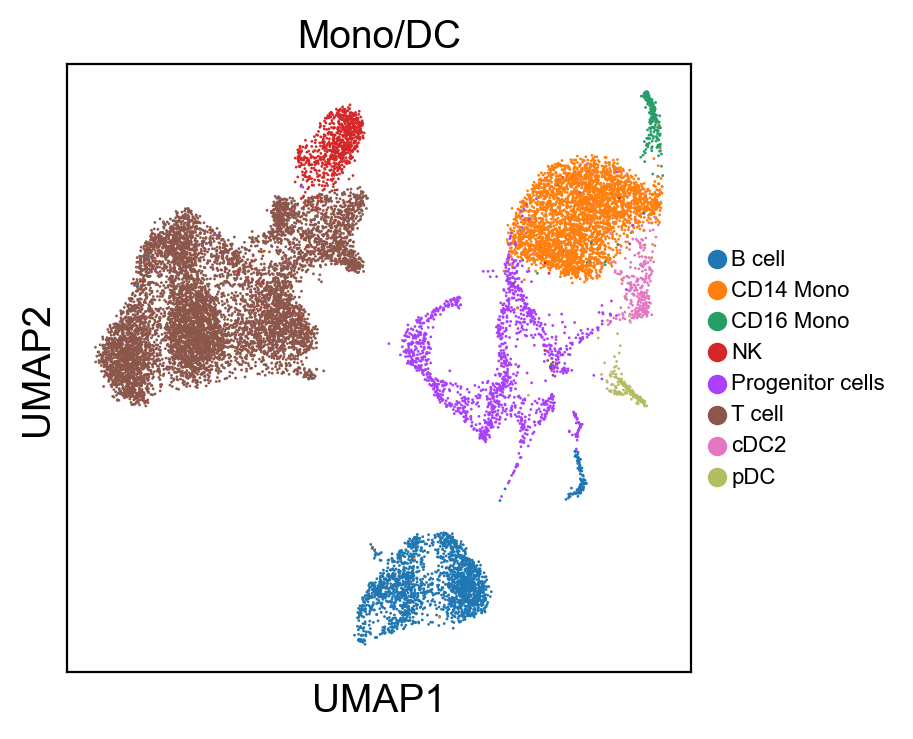
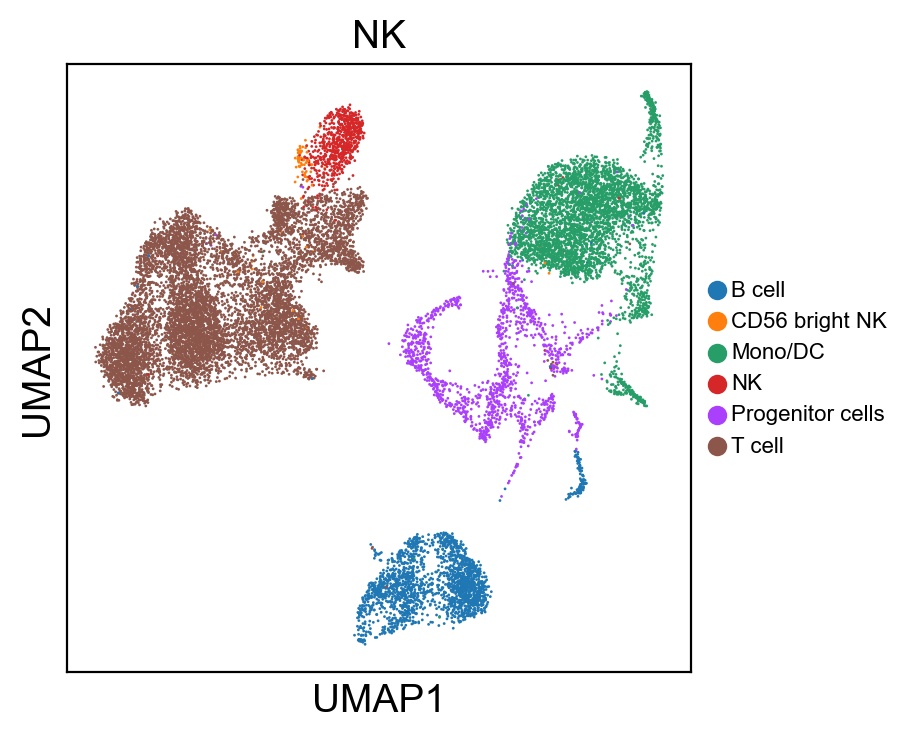
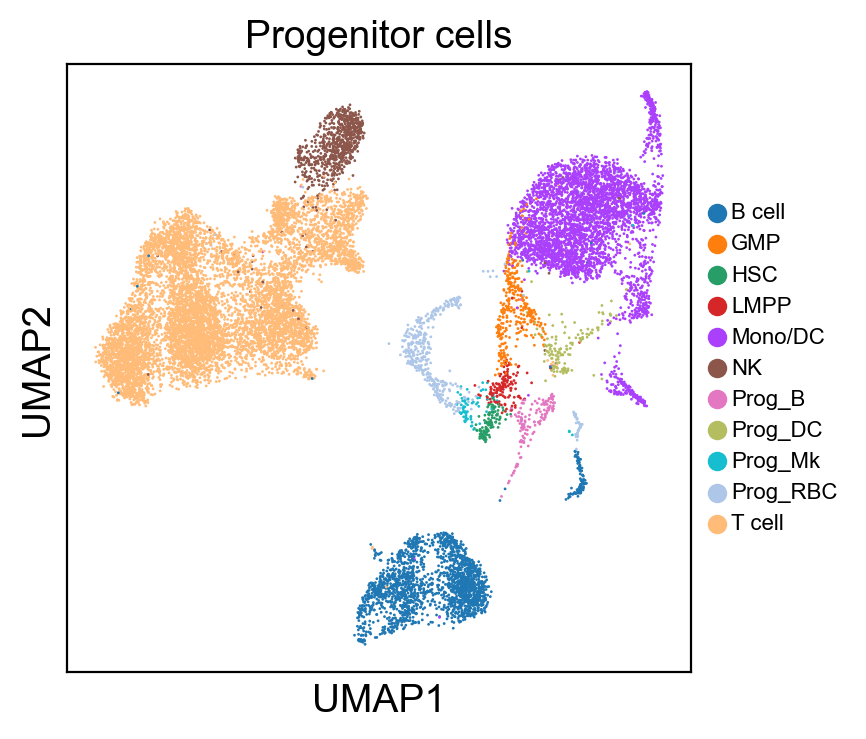
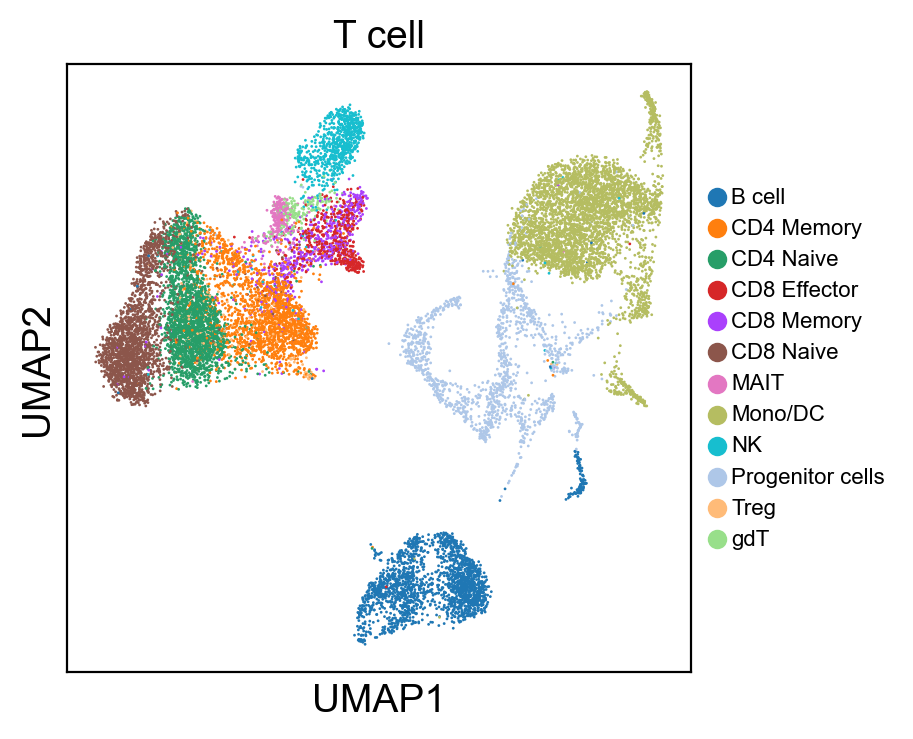
[36]:
sc.pl.umap(rna_adata, color=['T cell'], legend_loc=None, legend_fontsize=8., size=4.)
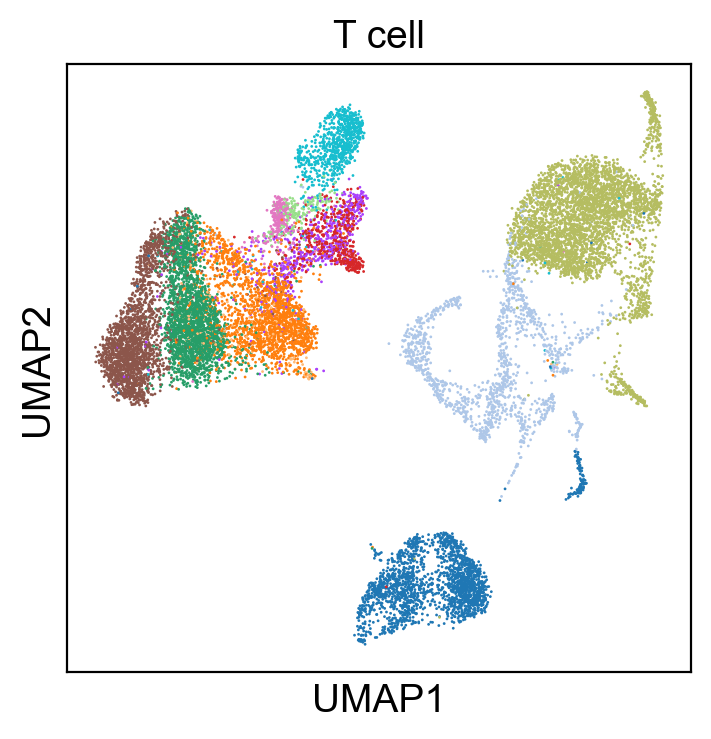
Compare Protein Profile with Gene Expression Profile
[37]:
rna_adata.var_names[rna_adata.var_names.str.contains('ITGA')]
[37]:
Index(['ITGA10', 'ITGA6', 'ITGA4', 'ITGAV', 'ITGA9', 'ITGA1', 'ITGA2', 'ITGA8',
'ITGA5', 'ITGA7', 'ITGAL', 'ITGAM', 'ITGAX', 'ITGAE', 'ITGA2B',
'ITGA3'],
dtype='object')
[38]:
coding_genes = {
'CD11a': ['ITGAL'],
'CD11c': ['ITGAX'],
'CD123': ['IL3RA'],
'CD127-IL7Ra': ['IL7R'],
'CD14': ['CD14'],
'CD16': ['FCGR3A', 'FCGR3B'],
'CD161': ['KLRB1'],
'CD19': ['CD19'],
'CD197-CCR7': ['CCR7'],
'CD25': ['IL2RA'],
'CD27': ['CD27'],
'CD278-ICOS': ['ICOS'],
'CD28': ['CD28'],
'CD3': ['CD3D', 'CD3E', 'CD3G'],
'CD34': ['CD34'],
'CD38': ['CD38'],
'CD4': ['CD4'],
'CD45RA': ['PTPRC'],
'CD45RO': ['PTPRC'],
'CD56': ['NCAM1'],
'CD57': ['B3GAT1'],
'CD69': ['CD69'],
'CD79b': ['CD79B'],
'CD8a': ['CD8A'],
'HLA.DR': ['HLA-DRA', 'HLA-DRB1']
}
[39]:
sc.settings.set_figure_params(dpi=50, facecolor='white')
temp_adata = adt_adata.copy()
for i in coding_genes:
for j in coding_genes[i]:
temp_adata.obs['RNA_' + j] = rna_adata[:, j].X.squeeze().tolist()
sc.pl.umap(temp_adata, color=[i] + ['RNA_' + j for j in coding_genes[i]], legend_fontsize=8., size=4.)
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1094: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if not is_categorical(df_full[k]):
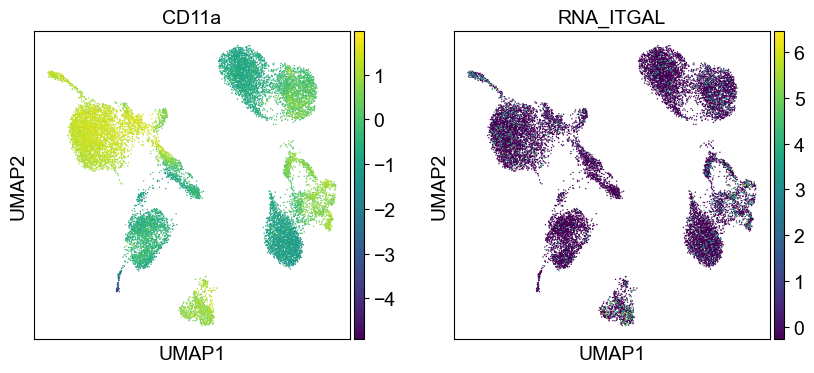
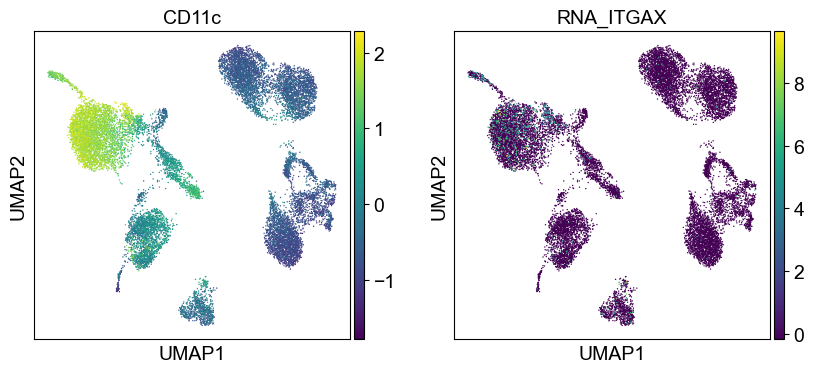
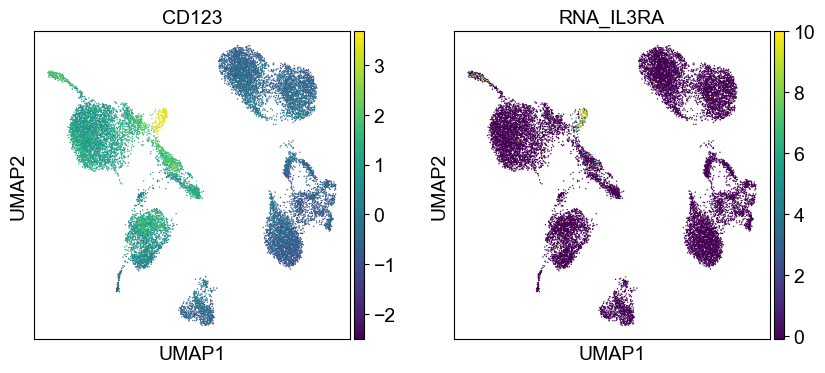
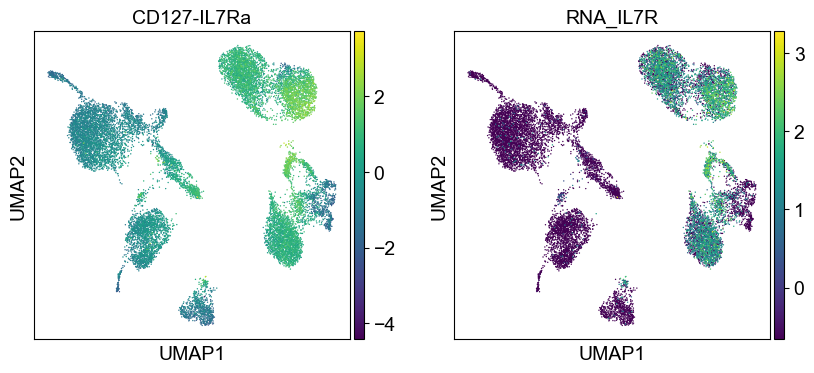
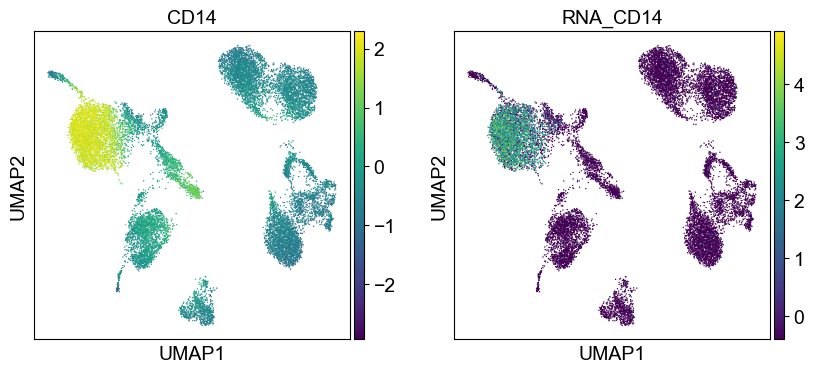
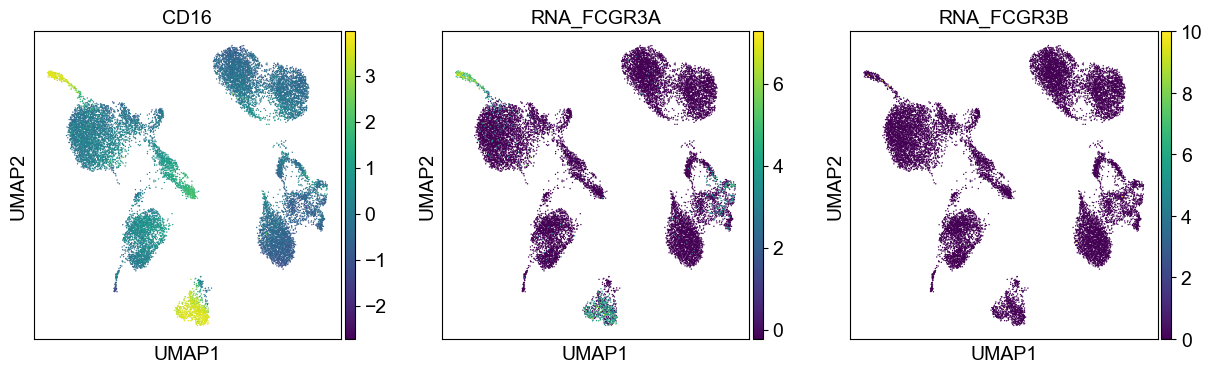
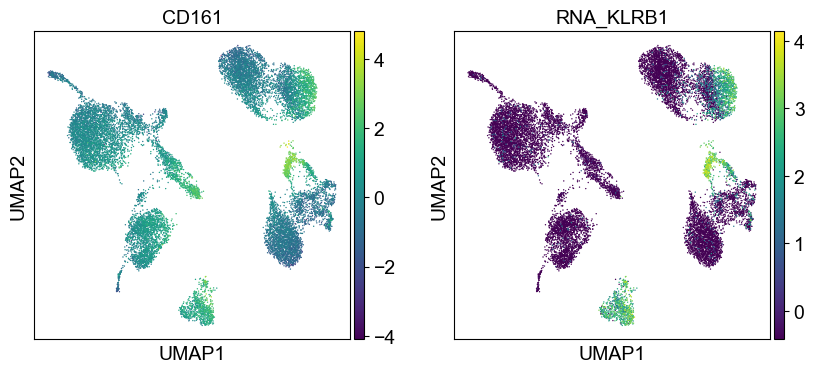
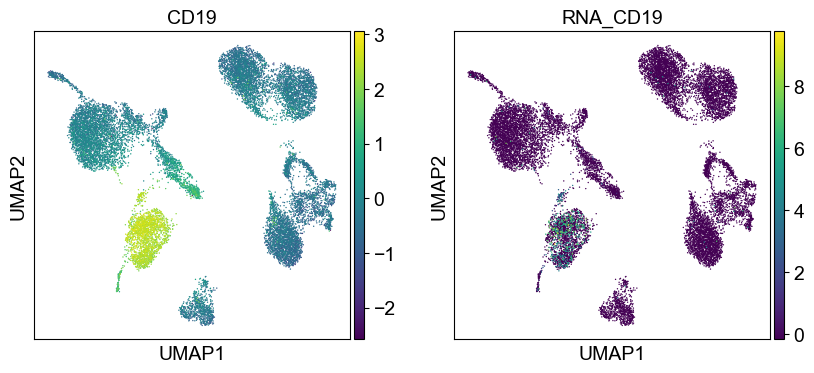
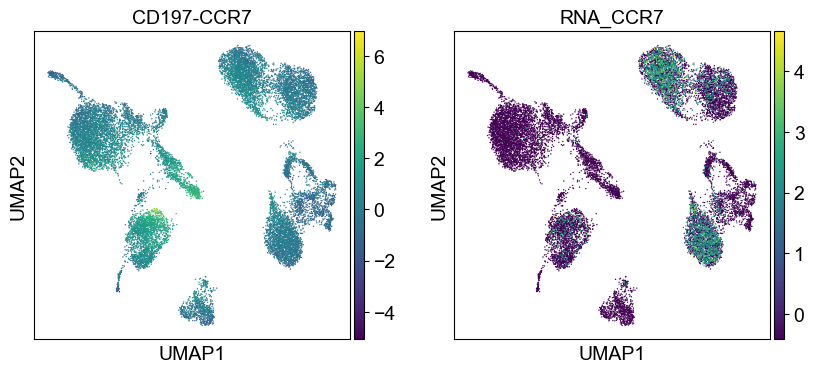
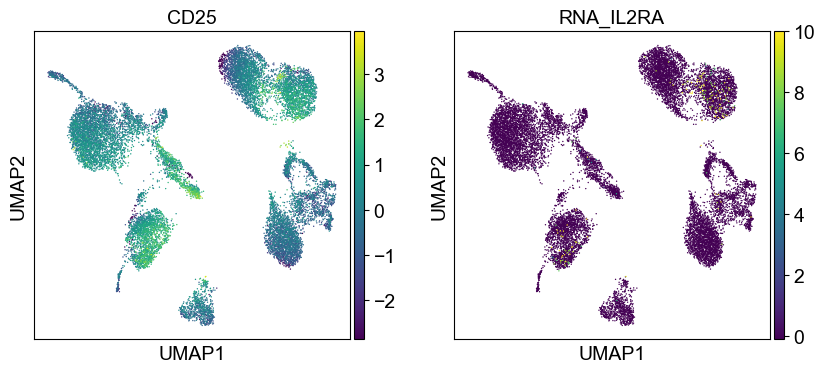
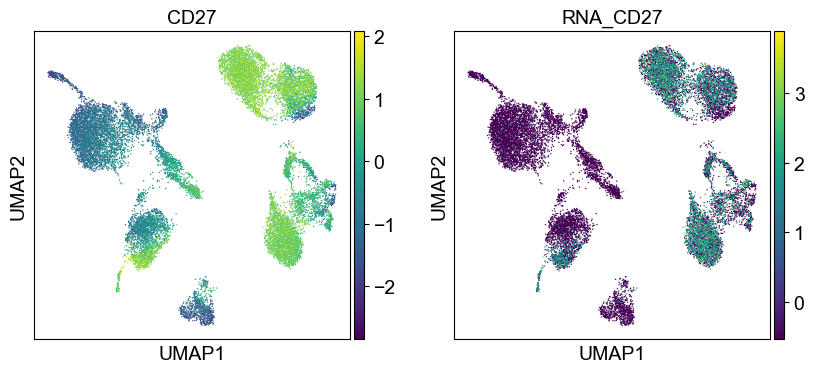
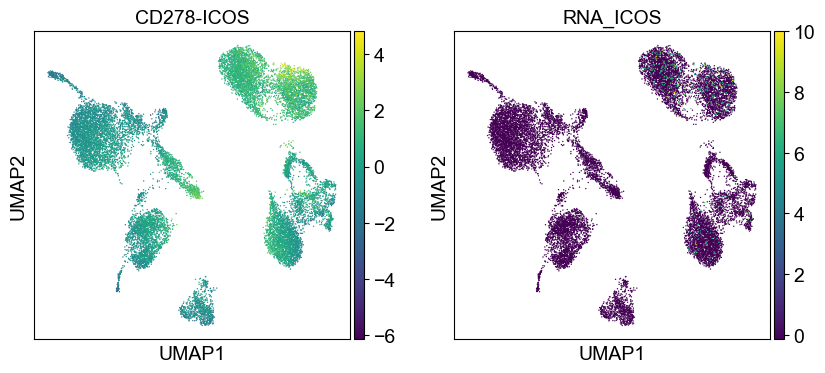
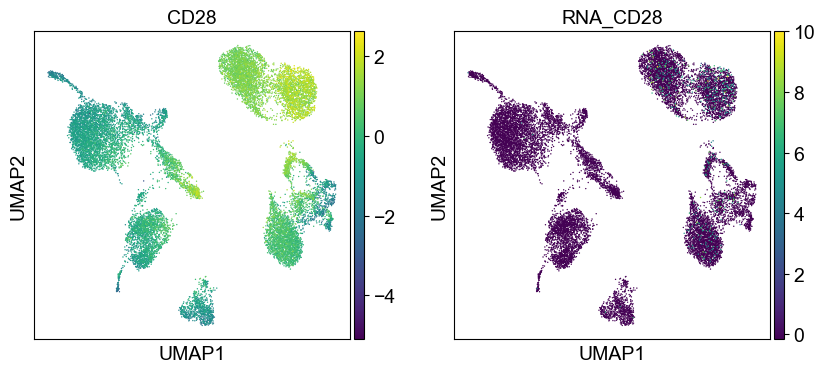
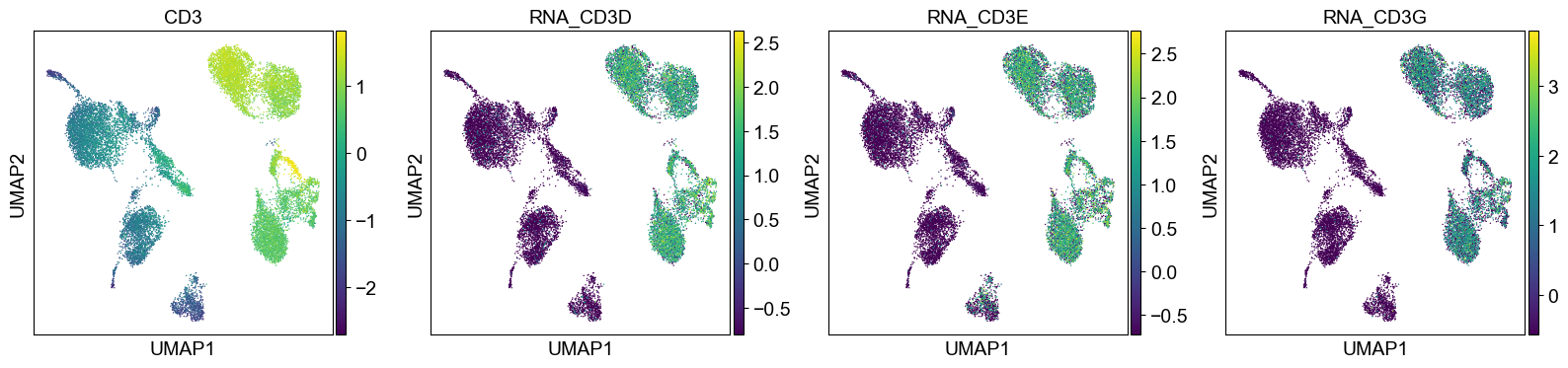
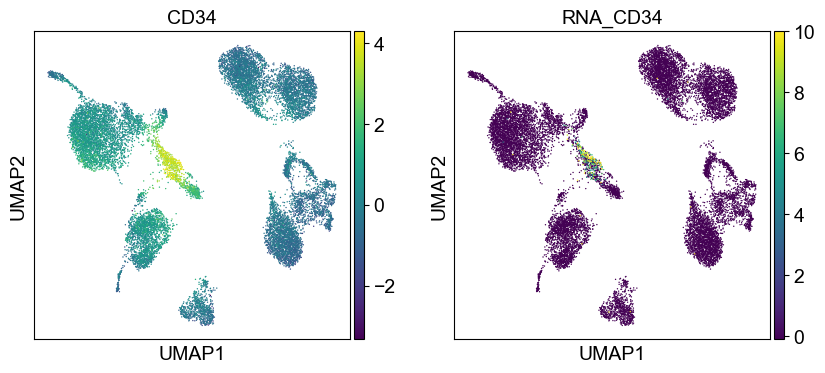
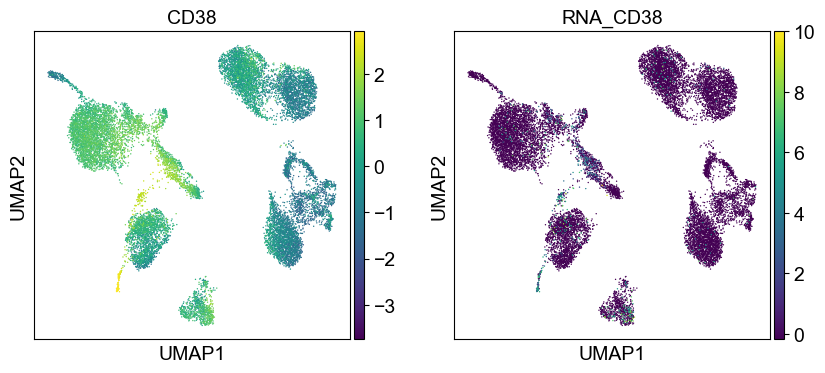
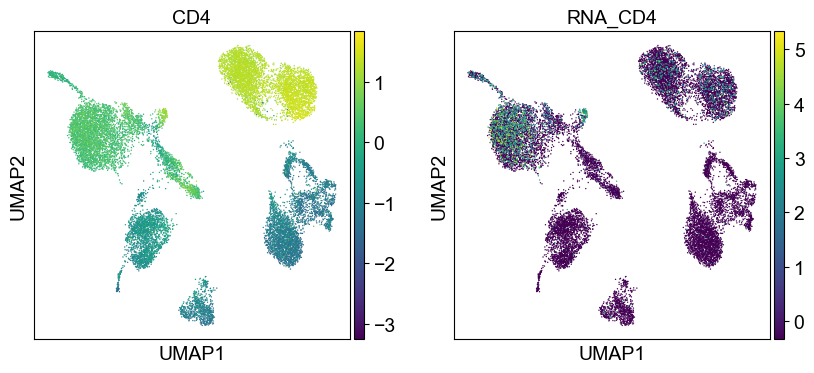
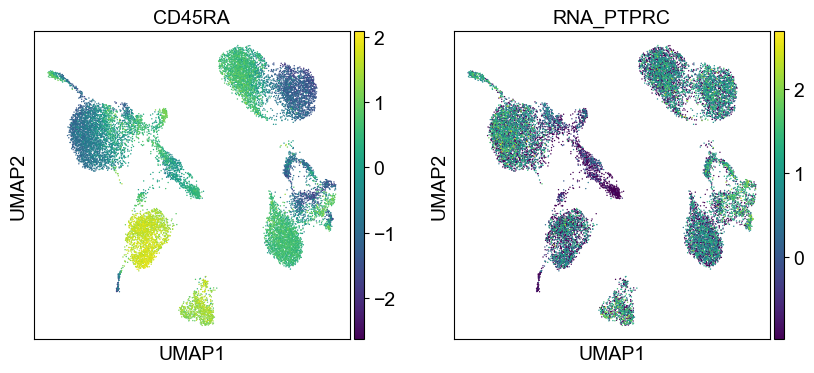
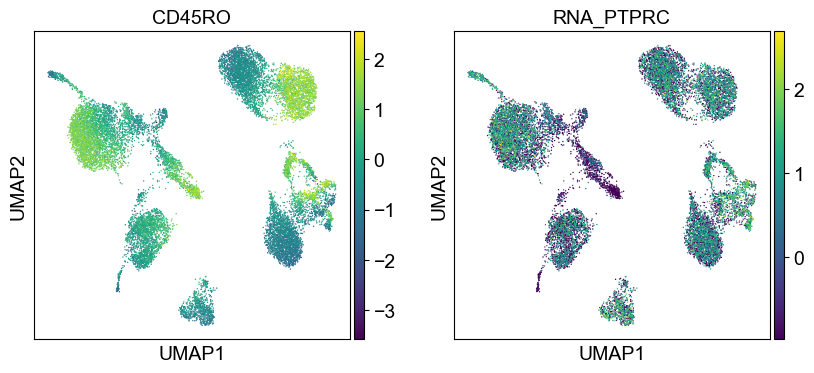
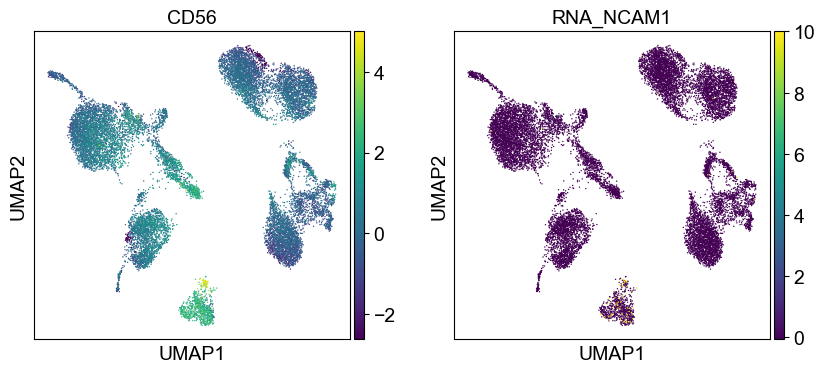
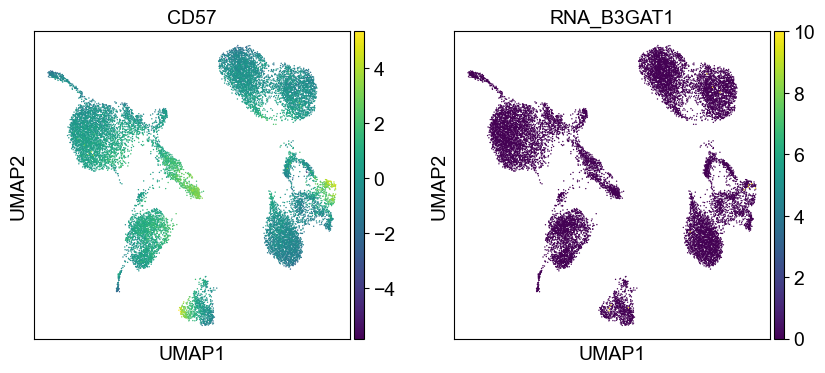
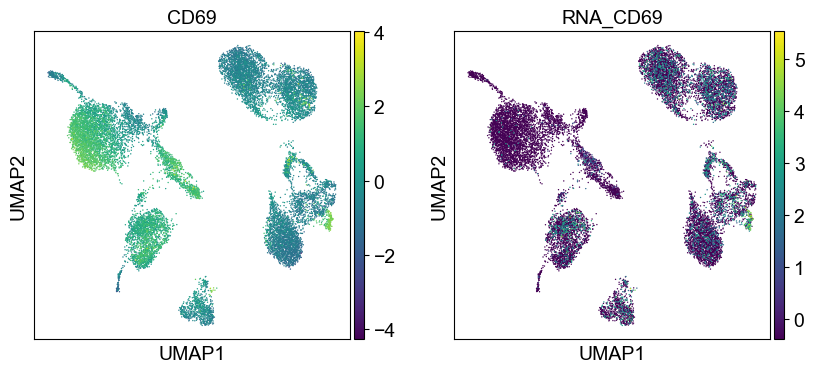
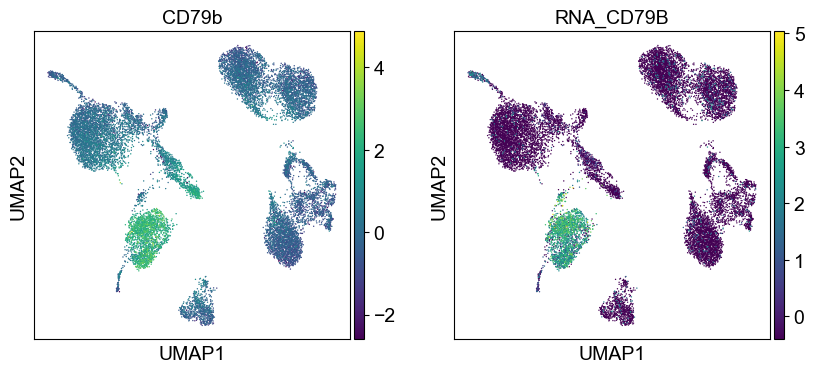
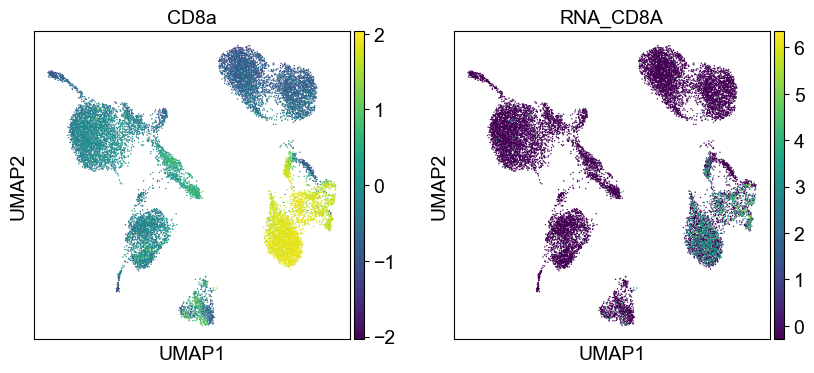
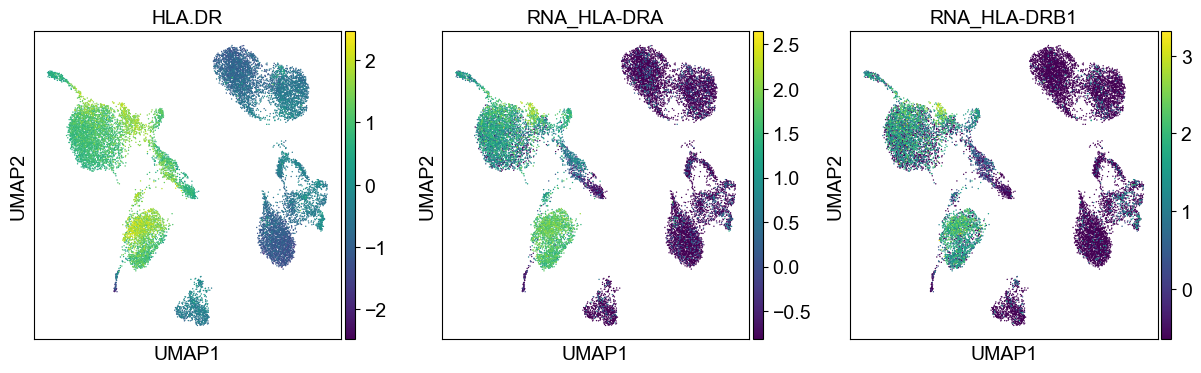
Use genes encoding these proteins for embedding
[40]:
coding_adata = rna_adata.copy()[:, sum(coding_genes.values(), [])]
coding_adata
[40]:
View of AnnData object with n_obs × n_vars = 16204 × 29
obs: 'Unnamed: 0', 'orig.ident', 'nCount_RNA', 'nFeature_RNA', 'nCount_ADT', 'nFeature_ADT', 'lane', 'donor', 'celltype.l1', 'celltype.l2', 'RNA.weight', 'celltype.l1.5', 'B cell', 'Mono/DC', 'NK', 'Progenitor cells', 'T cell', 'celltype.oi'
var: 'mean', 'std'
uns: 'log1p', 'pca', 'neighbors', 'umap', 'celltype.oi_colors', 'celltype.l1.5_colors', 'B cell_colors', 'Mono/DC_colors', 'NK_colors', 'Progenitor cells_colors', 'T cell_colors'
obsm: 'X_pca', 'X_umap'
varm: 'PCs'
obsp: 'distances', 'connectivities'
[41]:
sc.tl.pca(coding_adata, svd_solver='arpack')
computing PCA
with n_comps=28
Variable names are not unique. To make them unique, call `.var_names_make_unique`.
Variable names are not unique. To make them unique, call `.var_names_make_unique`.
finished (0:00:00)
[42]:
sc.settings.set_figure_params(dpi=100, facecolor='white')
sc.pl.pca_variance_ratio(coding_adata)
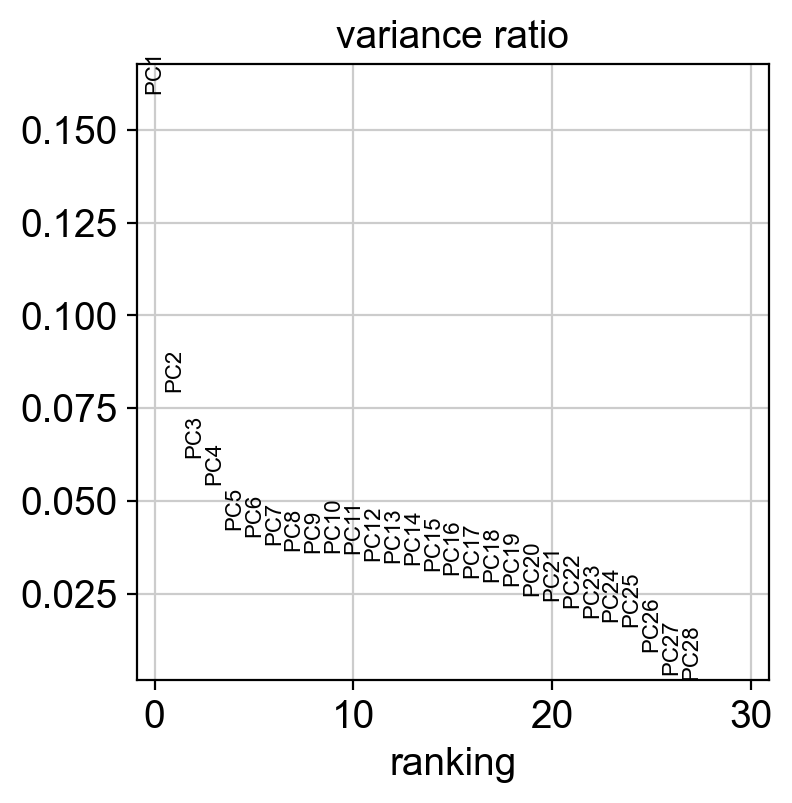
[43]:
sc.pp.neighbors(coding_adata, n_pcs=5, use_rep="X_pca")
sc.tl.umap(coding_adata)
computing neighbors
finished: added to `.uns['neighbors']`
`.obsp['distances']`, distances for each pair of neighbors
`.obsp['connectivities']`, weighted adjacency matrix (0:00:01)
computing UMAP
finished: added
'X_umap', UMAP coordinates (adata.obsm) (0:00:09)
[44]:
sc.pl.umap(coding_adata, color=['celltype.l1.5'], legend_loc="on data", legend_fontsize=8., size=5.)
sc.pl.umap(coding_adata, color=['celltype.l1.5'], legend_loc=None, legend_fontsize=8., size=5.)
sc.pl.umap(coding_adata, color=['celltype.l1.5'], legend_fontsize=8., size=5.)
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1192: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if is_string_dtype(df[key]) and not is_categorical(df[key])
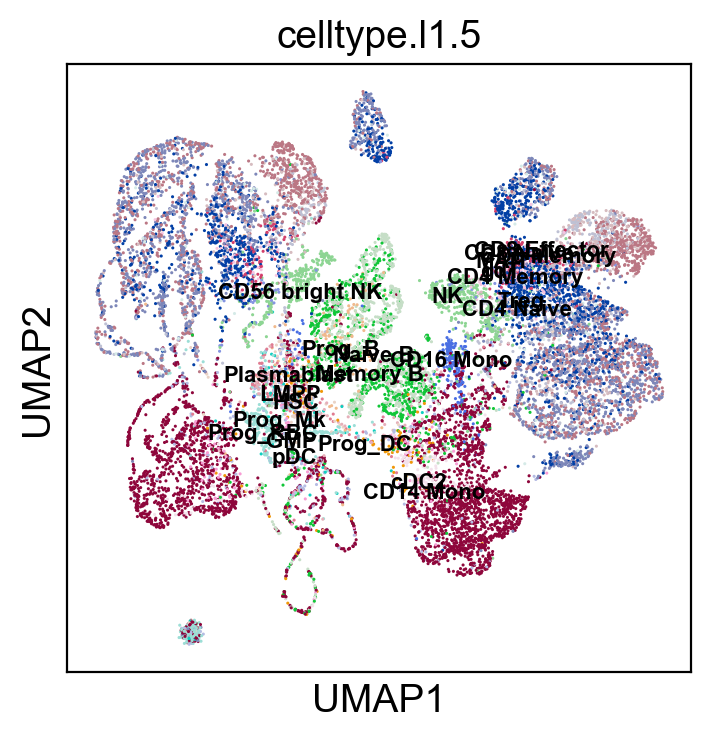
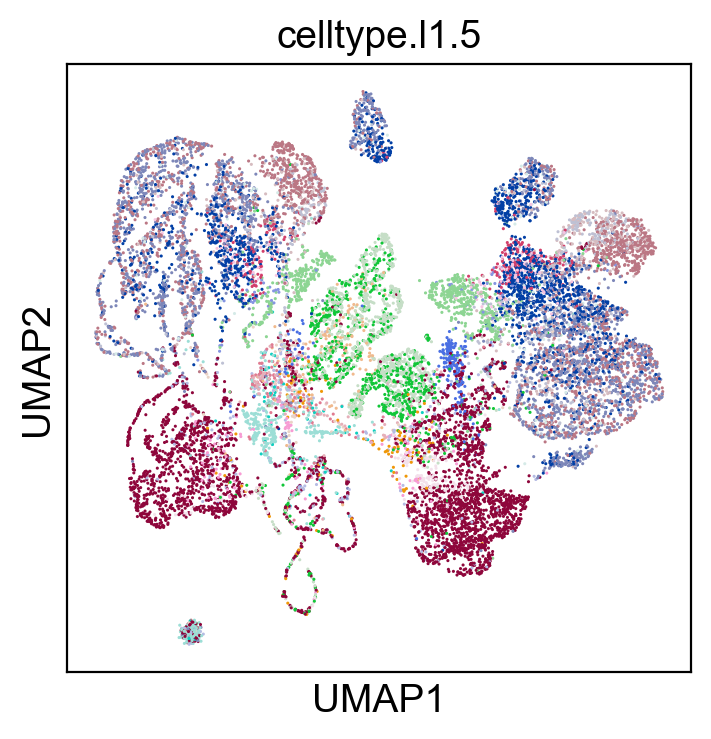
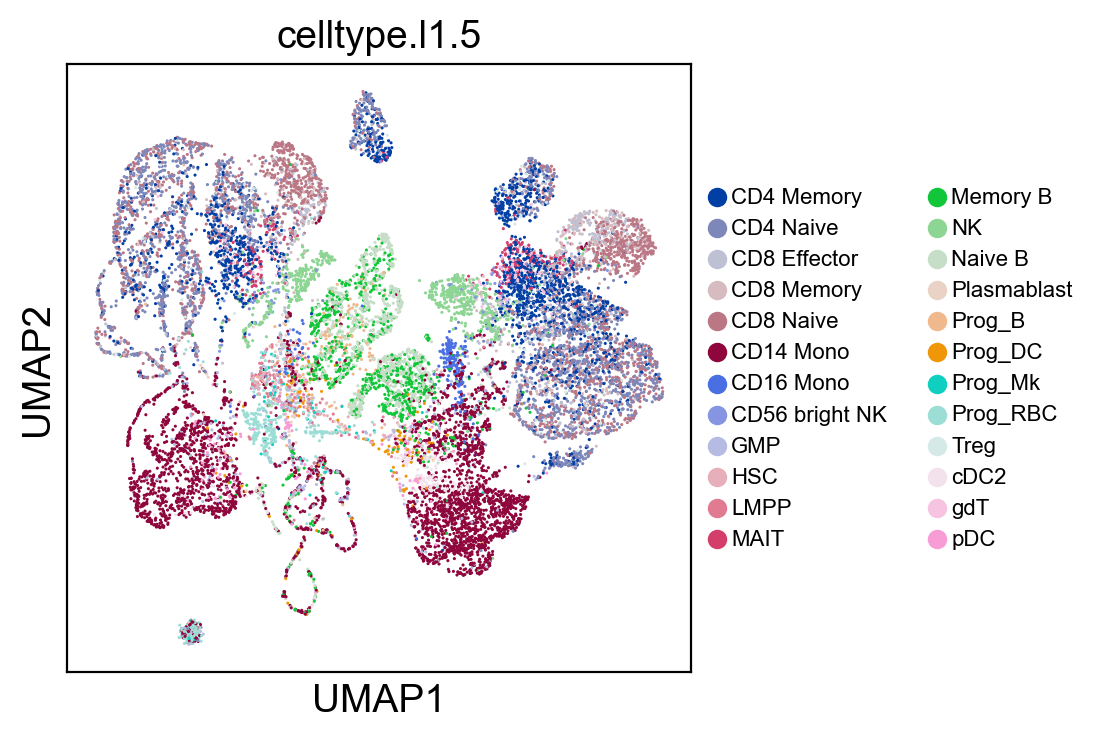
Validation
[46]:
w_df = pd.read_csv("bmnc-140.csv", index_col=0)
[47]:
new_adata = rna_adata.copy()[:, w_df.index.tolist()]
sc.tl.pca(new_adata, svd_solver='arpack')
new_adata
computing PCA
with n_comps=50
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1094: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if not is_categorical(df_full[k]):
finished (0:00:00)
[47]:
AnnData object with n_obs × n_vars = 16204 × 140
obs: 'Unnamed: 0', 'orig.ident', 'nCount_RNA', 'nFeature_RNA', 'nCount_ADT', 'nFeature_ADT', 'lane', 'donor', 'celltype.l1', 'celltype.l2', 'RNA.weight', 'celltype.l1.5', 'B cell', 'Mono/DC', 'NK', 'Progenitor cells', 'T cell', 'celltype.oi'
var: 'mean', 'std'
uns: 'log1p', 'pca', 'neighbors', 'umap', 'celltype.oi_colors', 'celltype.l1.5_colors', 'B cell_colors', 'Mono/DC_colors', 'NK_colors', 'Progenitor cells_colors', 'T cell_colors'
obsm: 'X_pca', 'X_umap'
varm: 'PCs'
obsp: 'distances', 'connectivities'
[48]:
sc.pl.pca_variance_ratio(new_adata)
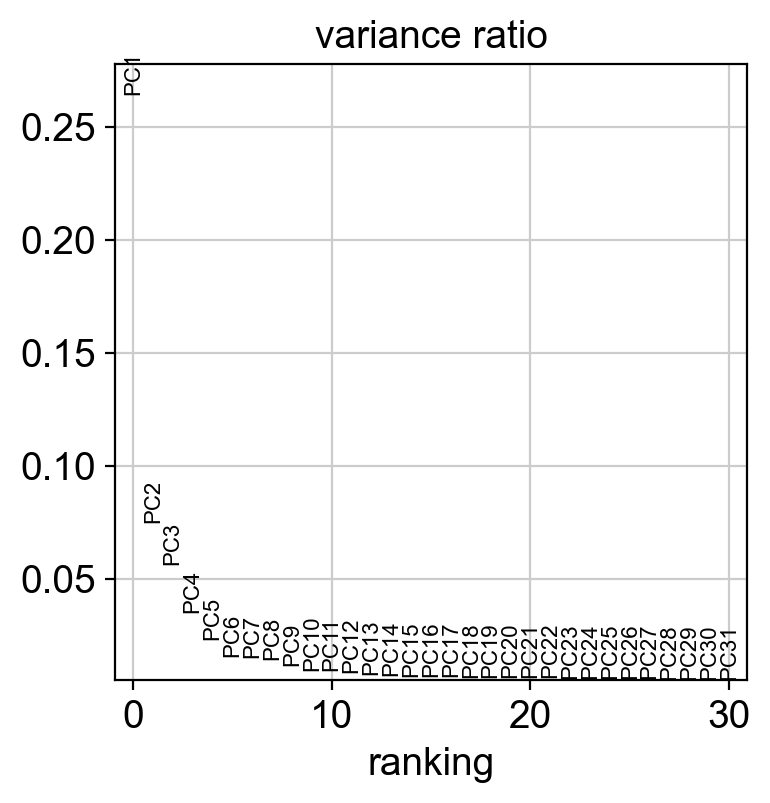
[57]:
sc.pp.neighbors(new_adata, n_pcs=14, use_rep="X_pca")
sc.tl.umap(new_adata, random_state=2)
computing neighbors
finished: added to `.uns['neighbors']`
`.obsp['distances']`, distances for each pair of neighbors
`.obsp['connectivities']`, weighted adjacency matrix (0:00:02)
computing UMAP
finished: added
'X_umap', UMAP coordinates (adata.obsm) (0:00:09)
[58]:
sc.pl.umap(new_adata, color=['celltype.oi'], groups=celltype_oi, legend_loc=None, legend_fontsize=8., size=5.)
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1192: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if is_string_dtype(df[key]) and not is_categorical(df[key])

[59]:
sc.pl.umap(new_adata, color=['celltype.l1.5'], legend_loc="on data", legend_fontsize=8., size=5.)
sc.pl.umap(new_adata, color=['celltype.l1.5'], legend_loc=None, legend_fontsize=8., size=5.)
sc.pl.umap(new_adata, color=['celltype.l1.5'], legend_fontsize=8., size=5.)
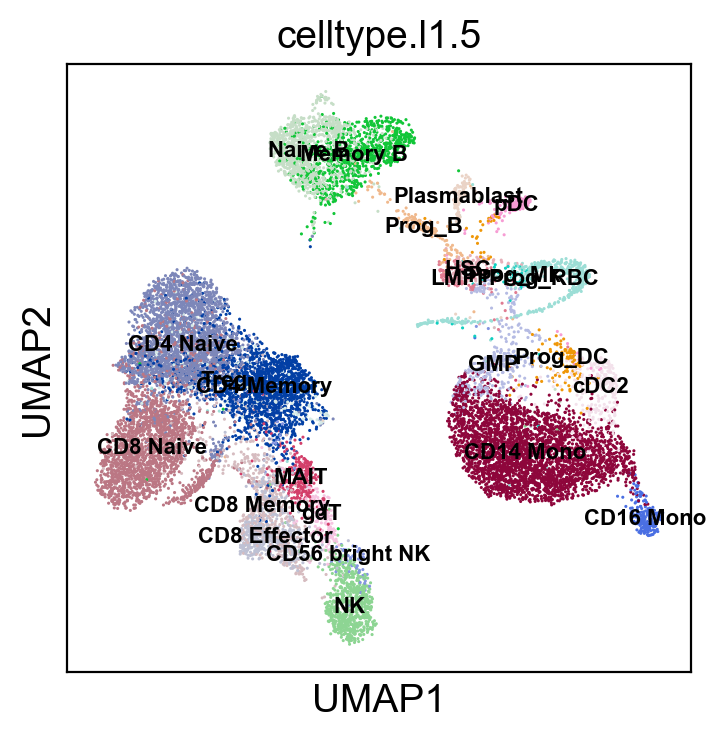
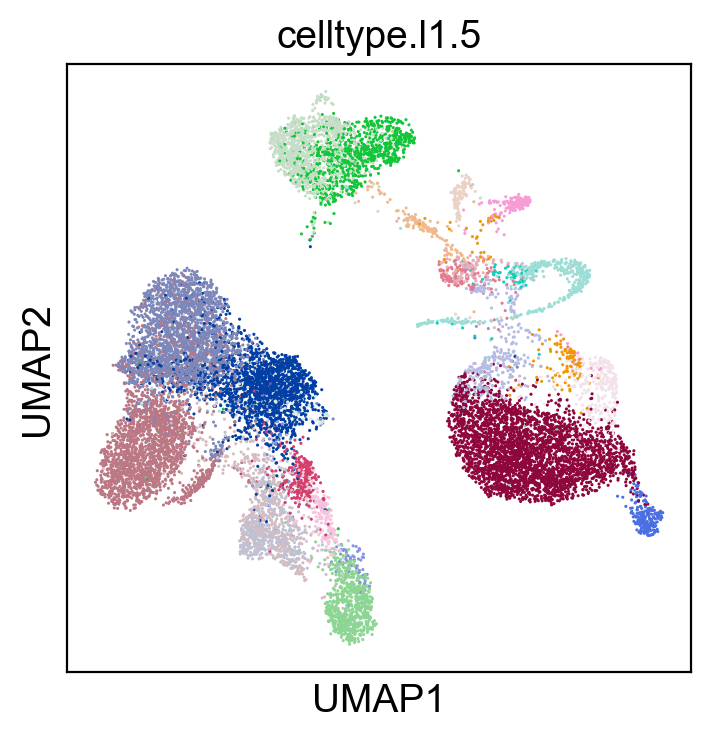
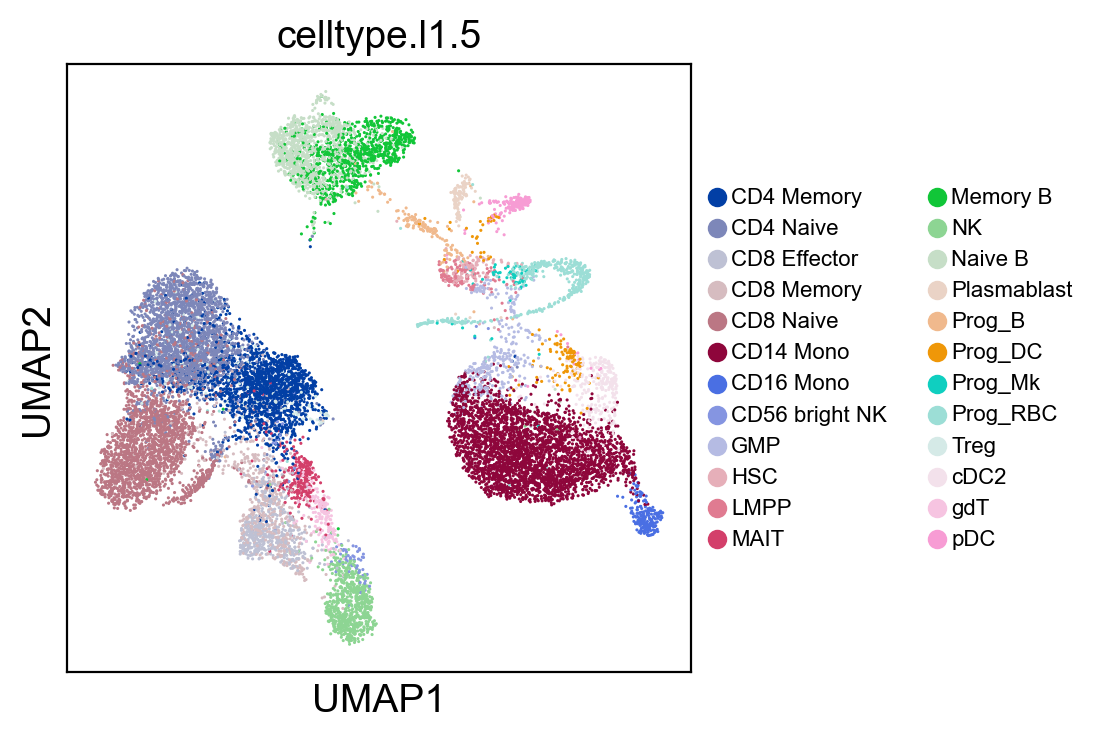
[52]:
sc.pl.umap(new_adata, color=['T cell'], legend_loc=None, legend_fontsize=8., size=4.)
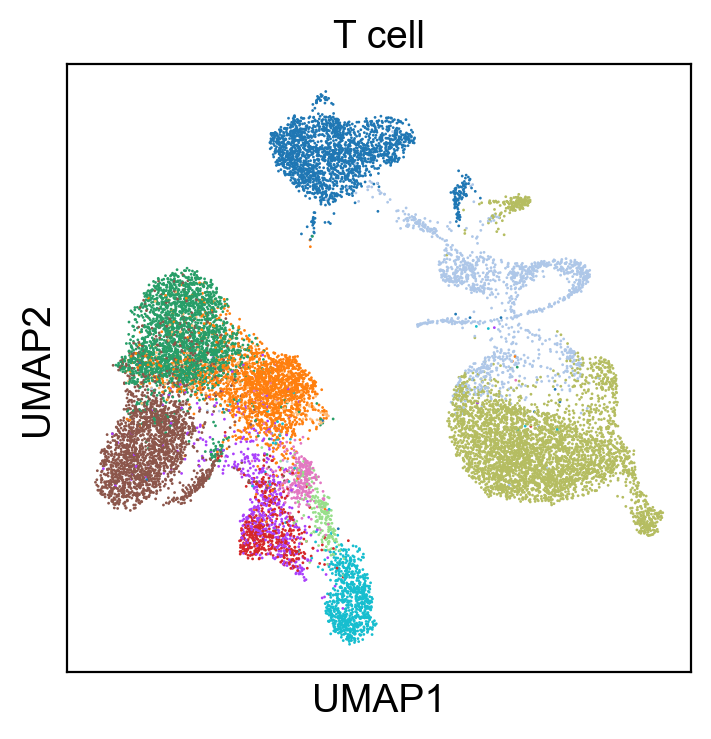
[53]:
for g in groups:
sc.pl.umap(new_adata, color=[g], legend_fontsize=8., size=4.)
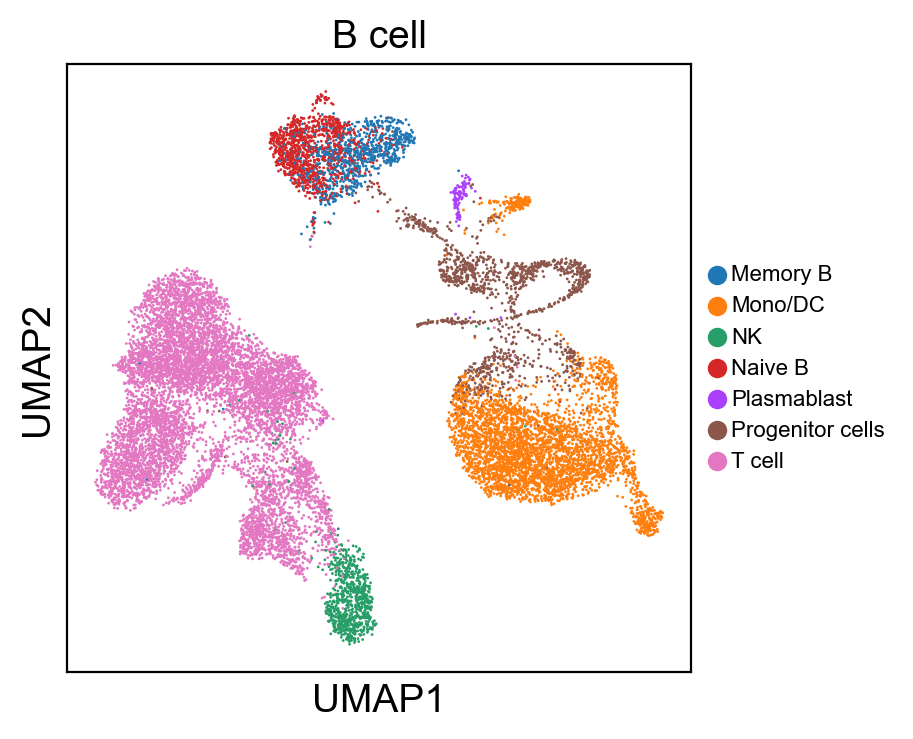
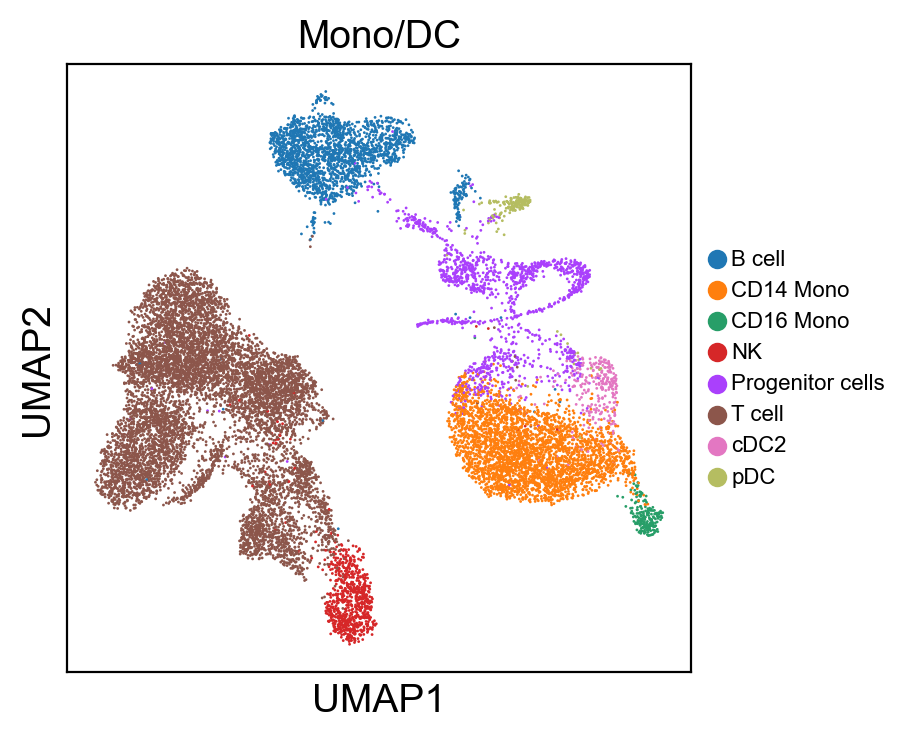
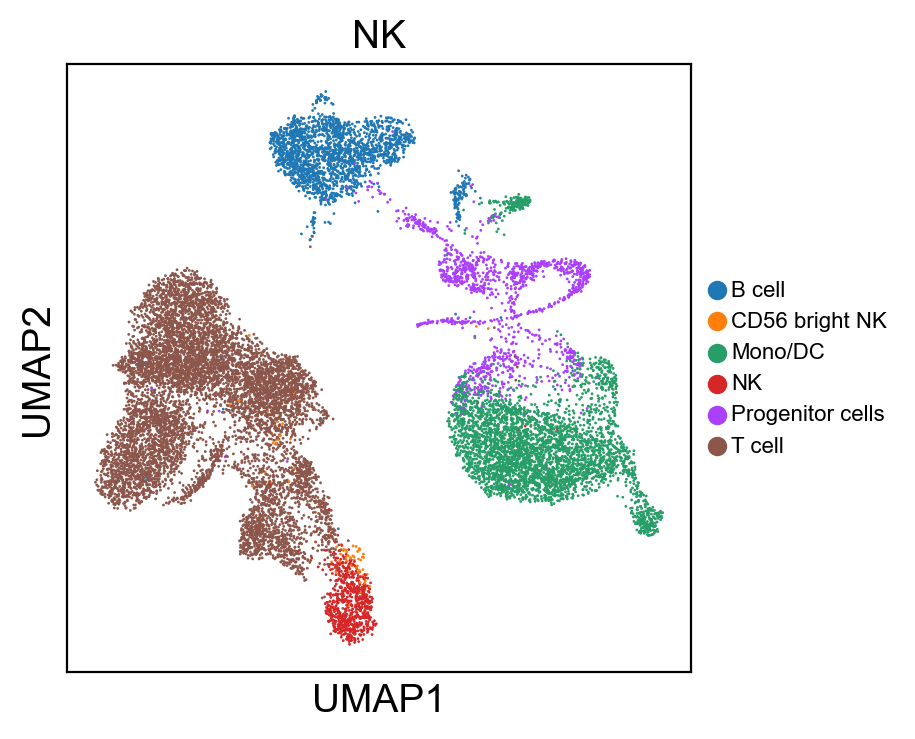
[ ]: