Bone Marrow Immune Cell Differentiation
[1]:
import scanpy as sc
import os
import pandas as pd
import numpy as np
import pickle as pkl
import matplotlib as mpl
import matplotlib.pyplot as plt
import scipy.stats
from scipy.io import mmread
sc.settings.verbosity = 3
[2]:
read_from_h5ad = True
h5ad_path = '../../AML van Galen/BM1234.h5ad'
if read_from_h5ad:
adata = sc.read_h5ad(h5ad_path)
else:
dem_names = ['GSM3587996_BM1.dem.txt.gz',
'GSM3587997_BM2.dem.txt.gz',
'GSM3587998_BM3.dem.txt.gz',
'GSM3588000_BM4.dem.txt.gz',
'GSM3588002_BM5-34p.dem.txt.gz',
'GSM3588003_BM5-34p38n.dem.txt.gz']
anno_names = ['GSM3587996_BM1.anno.txt.gz',
'GSM3587997_BM2.anno.txt.gz',
'GSM3587999_BM3.anno.txt.gz',
'GSM3588001_BM4.anno.txt.gz',
'GSM3588002_BM5-34p.anno.txt.gz',
'GSM3588003_BM5-34p38n.anno.txt.gz']
adata_list = [sc.read_text('../../AML van Galen/' + dem_names[i]).T for i in range(6)]
for i in range(6):
adata_list[i].obs = pd.read_table('../../AML van Galen/' + anno_names[i], index_col=0)
adata = adata_list[0].concatenate(adata_list[1:])
adata.write_h5ad(h5ad_path)
[3]:
adata
[3]:
AnnData object with n_obs × n_vars = 7698 × 27899
obs: 'NumberOfReads', 'AlignedToGenome', 'AlignedToTranscriptome', 'TranscriptomeUMIs', 'NumberOfGenes', 'CyclingScore', 'CyclingBinary', 'MutTranscripts', 'WtTranscripts', 'PredictionRF2', 'PredictionRefined', 'CellType', 'Score_HSC', 'Score_Prog', 'Score_GMP', 'Score_ProMono', 'Score_Mono', 'Score_cDC', 'Score_pDC', 'Score_earlyEry', 'Score_lateEry', 'Score_ProB', 'Score_B', 'Score_Plasma', 'Score_T', 'Score_CTL', 'Score_NK', 'batch'
[4]:
sc.settings.set_figure_params(dpi=80, facecolor='white')
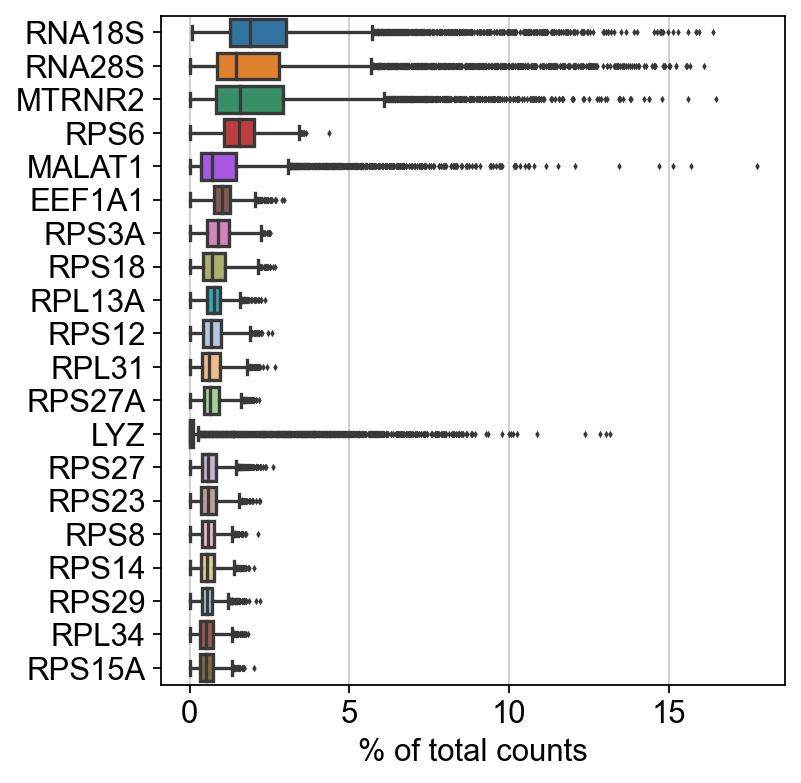
sc.pl.highest_expr_genes(adata, n_top=20)
normalizing counts per cell
finished (0:00:00)
[5]:
sc.pp.filter_cells(adata, min_genes=100)
sc.pp.filter_genes(adata, min_cells=3)
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1094: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if not is_categorical(df_full[k]):
filtered out 9444 genes that are detected in less than 3 cells
[6]:
sc.settings.set_figure_params(dpi=50, facecolor='white')
adata.var['mt'] = adata.var.index.str.startswith('MT-') # annotate the group of mitochondrial genes as 'mt'
sc.pp.calculate_qc_metrics(adata, qc_vars=['mt'], percent_top=None, log1p=False, inplace=True)
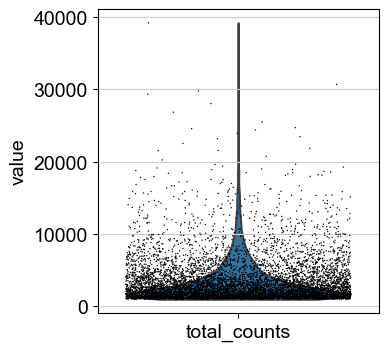
sc.pl.violin(adata, ['n_genes_by_counts'], jitter=0.4)
sc.pl.violin(adata, ['total_counts'], jitter=0.4)
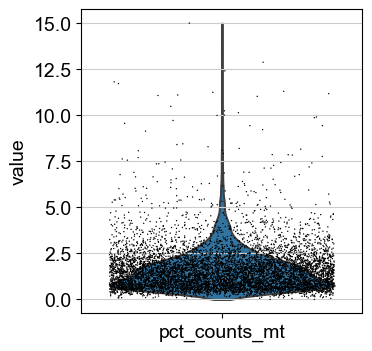
sc.pl.violin(adata, ['pct_counts_mt'], jitter=0.4)
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\seaborn\_decorators.py:43: FutureWarning: Pass the following variable as a keyword arg: x. From version 0.12, the only valid positional argument will be `data`, and passing other arguments without an explicit keyword will result in an error or misinterpretation.
FutureWarning
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\seaborn\_decorators.py:43: FutureWarning: Pass the following variable as a keyword arg: x. From version 0.12, the only valid positional argument will be `data`, and passing other arguments without an explicit keyword will result in an error or misinterpretation.
FutureWarning

C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\seaborn\_decorators.py:43: FutureWarning: Pass the following variable as a keyword arg: x. From version 0.12, the only valid positional argument will be `data`, and passing other arguments without an explicit keyword will result in an error or misinterpretation.
FutureWarning
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\seaborn\_decorators.py:43: FutureWarning: Pass the following variable as a keyword arg: x. From version 0.12, the only valid positional argument will be `data`, and passing other arguments without an explicit keyword will result in an error or misinterpretation.
FutureWarning
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\seaborn\_decorators.py:43: FutureWarning: Pass the following variable as a keyword arg: x. From version 0.12, the only valid positional argument will be `data`, and passing other arguments without an explicit keyword will result in an error or misinterpretation.
FutureWarning
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\seaborn\_decorators.py:43: FutureWarning: Pass the following variable as a keyword arg: x. From version 0.12, the only valid positional argument will be `data`, and passing other arguments without an explicit keyword will result in an error or misinterpretation.
FutureWarning
[7]:
sc.settings.set_figure_params(dpi=50, facecolor='white')
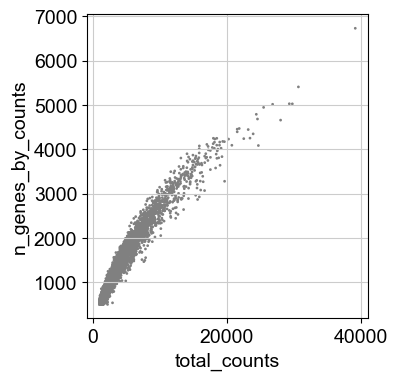
sc.pl.scatter(adata, x='total_counts', y='pct_counts_mt')
sc.pl.scatter(adata, x='total_counts', y='n_genes_by_counts')

[8]:
sc.pp.normalize_total(adata, target_sum=1e4)
sc.pp.log1p(adata)
normalizing counts per cell
finished (0:00:00)
[9]:
sc.pp.highly_variable_genes(adata)
sc.pl.highly_variable_genes(adata)
adata.var.highly_variable.sum()
extracting highly variable genes
finished (0:00:01)
--> added
'highly_variable', boolean vector (adata.var)
'means', float vector (adata.var)
'dispersions', float vector (adata.var)
'dispersions_norm', float vector (adata.var)
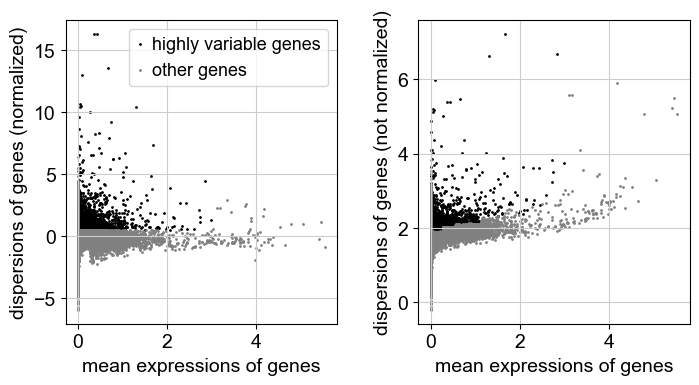
[9]:
3352
[10]:
adata.raw = adata
[11]:
adata = adata[:, adata.var.highly_variable]
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1094: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if not is_categorical(df_full[k]):
[12]:
sc.pp.scale(adata, max_value=10)
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\scanpy\preprocessing\_simple.py:806: UserWarning: Revieved a view of an AnnData. Making a copy.
view_to_actual(adata)
[13]:
sc.settings.set_figure_params(dpi=100, facecolor='white')
sc.tl.pca(adata, svd_solver='arpack')
sc.pl.pca(adata, color=['CellType'], size=5., frameon=False)
sc.pl.pca_variance_ratio(adata, log=True, n_pcs=50)
computing PCA
on highly variable genes
with n_comps=50
finished (0:00:02)
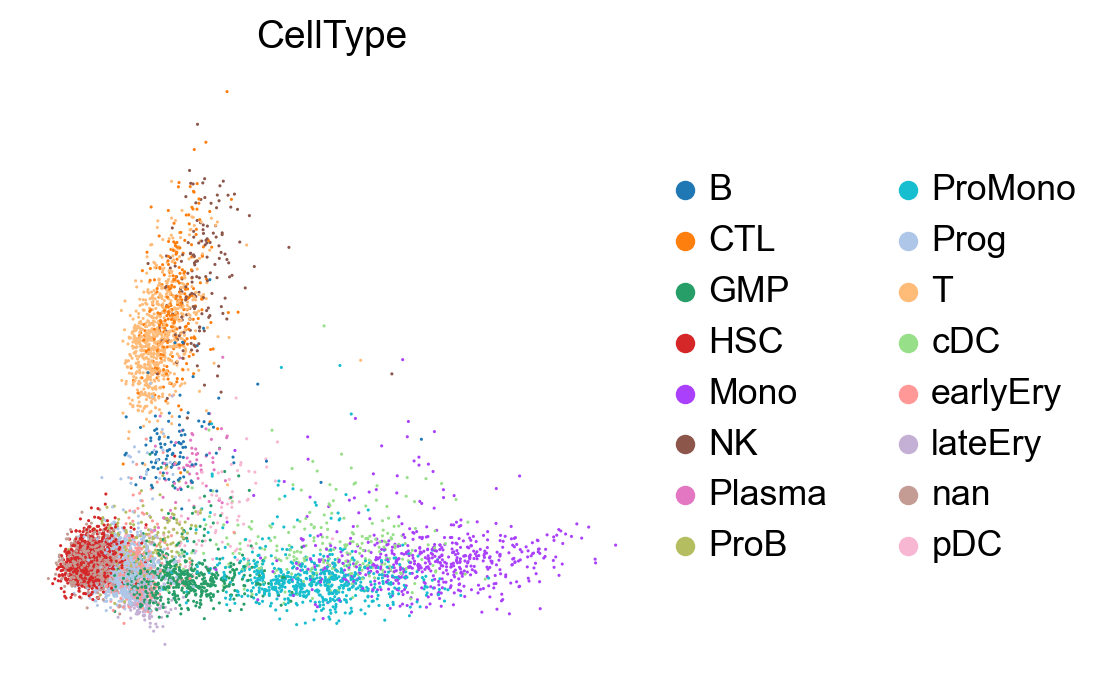
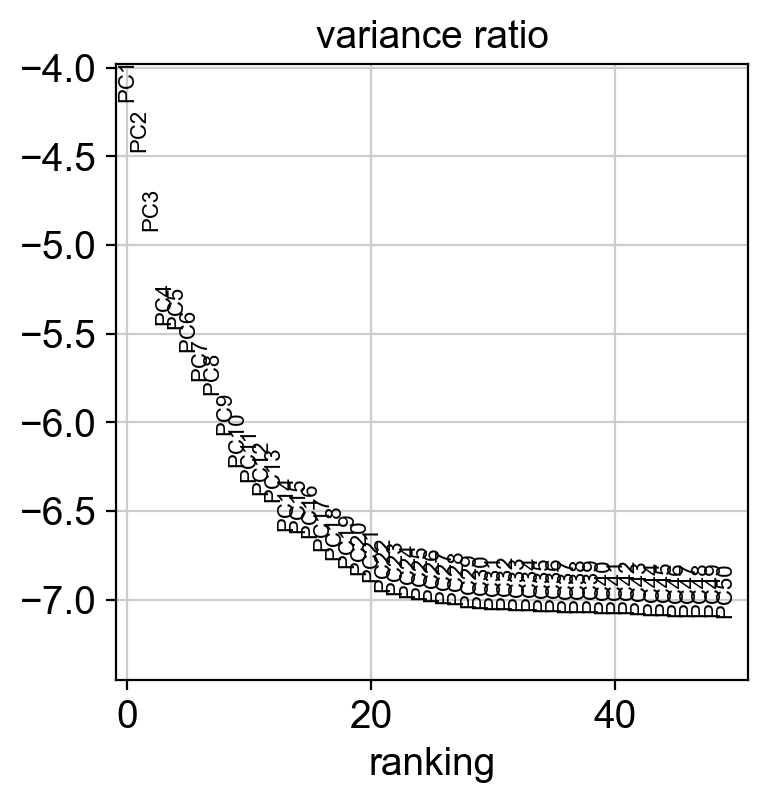
[14]:
sc.pp.neighbors(adata, n_pcs=25)
sc.tl.umap(adata)
sc.settings.set_figure_params(dpi=100, facecolor='white')
sc.pl.umap(adata, color=['CellType', 'batch'], legend_loc="on data", frameon=False, size=5., legend_fontsize=10, title='HVG')
computing neighbors
using 'X_pca' with n_pcs = 25
finished: added to `.uns['neighbors']`
`.obsp['distances']`, distances for each pair of neighbors
`.obsp['connectivities']`, weighted adjacency matrix (0:00:04)
computing UMAP
finished: added
'X_umap', UMAP coordinates (adata.obsm) (0:00:10)
WARNING: The title list is shorter than the number of panels. Using 'color' value instead for some plots.

[15]:
sc.pl.umap(adata, color=['CellType'], legend_loc=None, frameon=False, size=5., legend_fontsize=10, title='')

[16]:
sc.pl.umap(adata, color=['CellType'], frameon=False, size=5., legend_fontsize=10, title='')

SCMER Feature Selection
[17]:
import sys
sys.path.insert(0, '..')
import compactmarker
model = compactmarker.UmapL1(w=1., lasso=3.3e-4, ridge=0., n_pcs=25, perplexity=100., use_beta_in_Q=True, n_threads=6,
max_outer_iter=3)
model.fit(adata.X)
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 0.767693
Done. Elapsed time: 48.19 seconds. Total: 48.19 seconds.
Creating model without batches...
Optimizing using OWLQN (because lasso is nonzero)...
0 loss: 5.5735859870910645 Nonzero: 110 Elapsed time: 178.27 seconds. Total: 226.47 seconds.
1 loss: 3.7979912757873535 Nonzero: 110 Elapsed time: 183.35 seconds. Total: 409.81 seconds.
2 loss: 3.7917709350585938 Nonzero: 110 Elapsed time: 183.99 seconds. Total: 593.80 seconds.
final loss: 3.7223687171936035 Nonzero: 110 Elapsed time: 2.86 seconds. Total: 596.66 seconds.
[17]:
<compactmarker._umap_l1.UmapL1 at 0x28971188f88>
[18]:
selected_genes = adata.var_names[model.get_mask(100)]
print(*selected_genes)
print(len(selected_genes))
AHSP ANXA1 APOOL AREG ASPM ATP2B1 BCL11A C5AR1 CA1 CALR CCL5 CD3D CD52 CD74 CDK6 CENPF CENPU CLC CST3 CSTA CTSS CYBB DNTT DUSP1 EGR1 ELANE EREG ETS1 FCER1A FCN1 FGL2 FYB1 GNLY GPR183 GZMB HBA1 HBB HBD HIST1H1B HIST1H1C HIST1H2AJ HIST1H4C HLA-B HLA-DPA1 HLA-DRA HLA-E HLF HMGB2 HMGN2 HOPX HSP90B1 IGLL1 IL7R IQGAP1 IRF8 JCHAIN JUN KIAA0087 KLF6 MEIS1 MKI67 MNDA MPO MS4A1 MSI2 MT-ND5 MZB1 NAMPT NEAT1 NPR3 NRIP1 NUSAP1 PDZD8 PIK3R1 PRG2 PRKG2 PRSS2 PRTN3 PSAP PTPRC RNASE2 S100A10 S100A12 S100A6 SAMHD1 SAMSN1 SAT1 SLC40A1 SOX4 SPINK2 STK17B STMN1 TCF4 THBS1 TOP2A TXNDC5 TXNIP VCAN VPREB1 ZFAS1
100
Validation
[19]:
established_markers = {
"Undifferentiated": ["MEIS1", "EGR1", "MSI2", "CD38", "CD34", "PROM1", "EGFL7"],
"Myeloid GMP-Promono": ['MPO', "ELANE", "CTSG", "AZU1"],
"Myeloid Promono-differentiated": ["LYST", "LYZ", "CEBPD", "MNDA", "FCER1G", "CLEC4A"],
"Myeloid mono": ['FCN1', "CD14", "C5AR1"],
"Myeloid cDC": ['CLEC10A', "FCER1A"],
"Myeloid pDC": ['CLEC4C', 'PTPRS', 'IRF8', 'TCF4', 'JCHAIN', 'GZMB'],
"Ery": ['CSF1', "KIT", "HBB", "HBD", "GYPA"],
"Lymphoid Pro B": ['CD24', 'EBF1', 'MME', 'VPREB1', 'PAX5', 'CD19', 'CD79A'],
"Lymphoid B": ['MS4A1', 'BANK1'],
"Lymphoid Plasma": ['MZB1', 'IGLL5', 'JCHAIN'],
'Lymphoid T/CLT': ['CD3D', 'CD3G', 'IL32', 'IL7R', 'TCF7'],
'Lymphoid CLT/NK': ['CD8A', 'KLRB1', 'KLRD1', 'GZMB', 'NCAM1', 'CCL5', 'GZMK']
}
from IPython.display import Markdown, display
def printmd(string):
display(Markdown(string))
selected_markers = selected_genes
all_markers = adata.var_names.tolist()
for i in established_markers:
temp = " ".join((i, str(len(set(established_markers[i]).intersection(set(selected_markers)))),
'/', str(len(set(established_markers[i]).intersection(set(all_markers)))),
"(", str(len(established_markers[i])), "): "))
printmd(temp + '_' + '_, _'.join(set(established_markers[i]).intersection(set(selected_markers))) + '_')
Undifferentiated 3 / 3 ( 7 ): MEIS1, MSI2, EGR1
Myeloid GMP-Promono 2 / 4 ( 4 ): ELANE, MPO
Myeloid Promono-differentiated 1 / 4 ( 6 ): MNDA
Myeloid mono 2 / 3 ( 3 ): FCN1, C5AR1
Myeloid cDC 1 / 1 ( 2 ): FCER1A
Myeloid pDC 4 / 6 ( 6 ): TCF4, JCHAIN, IRF8, GZMB
Ery 2 / 3 ( 5 ): HBD, HBB
Lymphoid Pro B 1 / 7 ( 7 ): VPREB1
Lymphoid B 1 / 2 ( 2 ): MS4A1
Lymphoid Plasma 2 / 3 ( 3 ): JCHAIN, MZB1
Lymphoid T/CLT 2 / 5 ( 5 ): CD3D, IL7R
Lymphoid CLT/NK 2 / 7 ( 7 ): CCL5, GZMB
[20]:
temp = set(selected_markers.copy())
for i in established_markers:
temp -= set(established_markers[i])
printmd('_' + '_, _'.join(temp) + '_')
S100A10, HLA-DPA1, HLA-E, ETS1, FGL2, HOPX, SAMHD1, ANXA1, HIST1H2AJ, BCL11A, CENPU, APOOL, HIST1H4C, PRTN3, MKI67, HLF, CENPF, TOP2A, DUSP1, NEAT1, DNTT, PRKG2, HLA-DRA, HMGN2, ASPM, GPR183, EREG, CD52, KLF6, TXNIP, CALR, HSP90B1, CA1, AHSP, CYBB, HBA1, CST3, ZFAS1, CTSS, AREG, IGLL1, HIST1H1B, SAMSN1, S100A12, PTPRC, GNLY, CDK6, JUN, NRIP1, ATP2B1, NPR3, HMGB2, CLC, FYB1, NAMPT, CD74, STK17B, SOX4, RNASE2, STMN1, PRG2, PRSS2, THBS1, VCAN, IQGAP1, MT-ND5, SLC40A1, TXNDC5, PSAP, PIK3R1, HLA-B, SPINK2, HIST1H1C, CSTA, KIAA0087, SAT1, PDZD8, NUSAP1, S100A6
[21]:
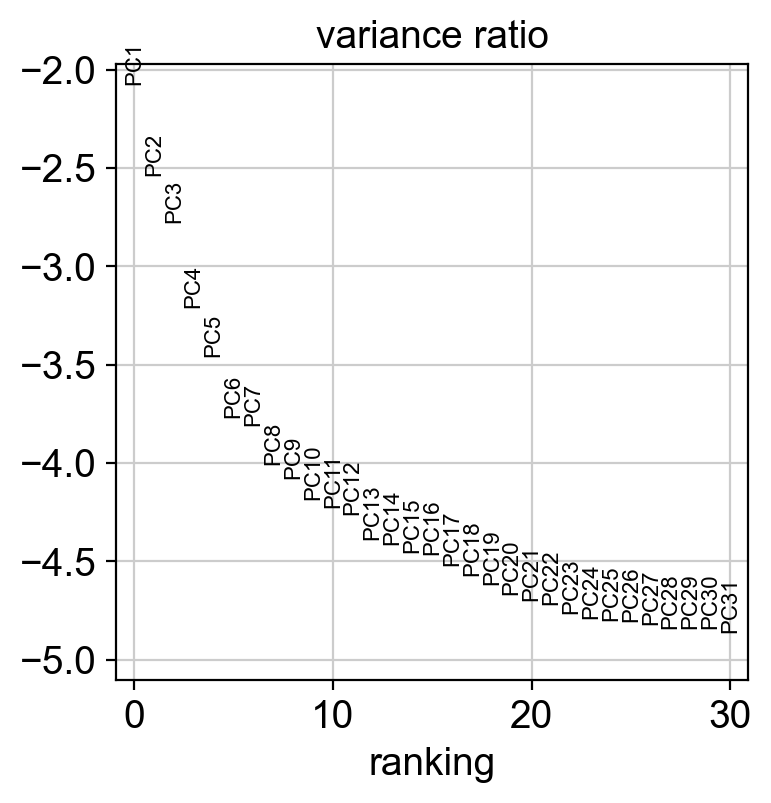
new_adata = adata[:, selected_genes]
sc.tl.pca(new_adata, svd_solver='arpack')
sc.pl.pca(new_adata, color=['CellType'])
sc.pl.pca_variance_ratio(new_adata, log=True)
computing PCA
on highly variable genes
with n_comps=50
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1094: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if not is_categorical(df_full[k]):
finished (0:00:00)
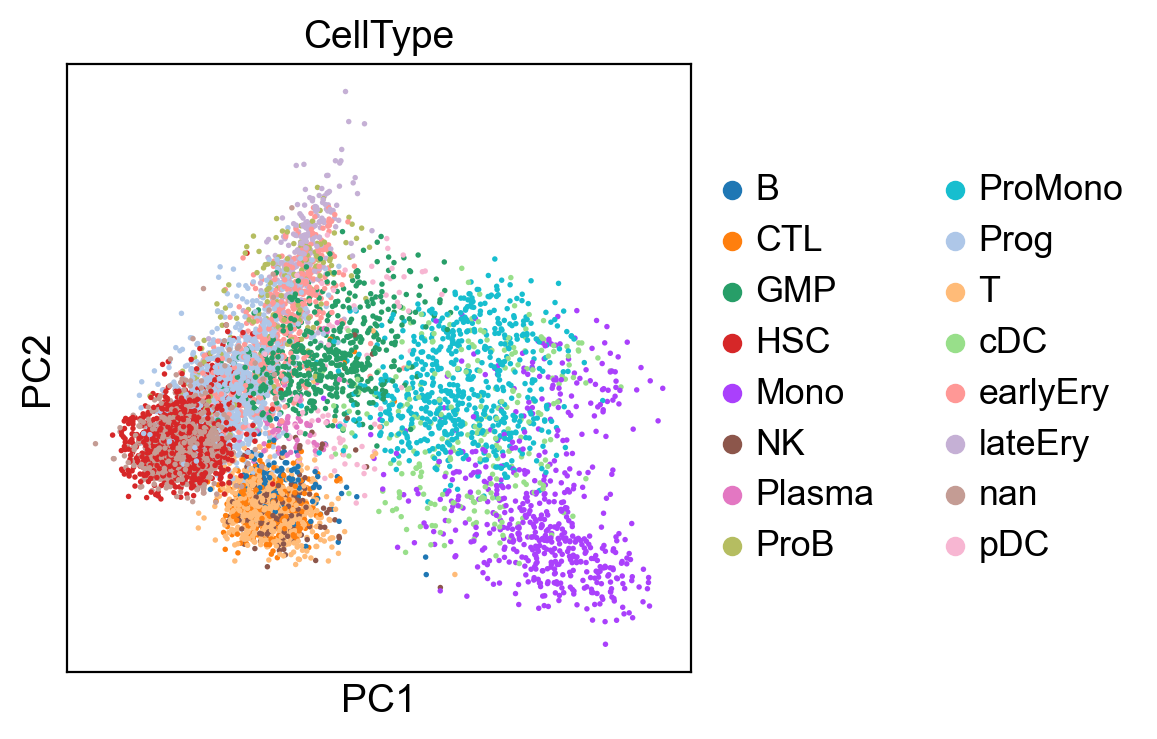
[22]:
sc.pp.neighbors(new_adata, n_pcs=12)
sc.tl.umap(new_adata)
sc.settings.set_figure_params(dpi=100, facecolor='white')
sc.pl.umap(new_adata, color=['CellType'], legend_loc="on data", frameon=False, size=5., legend_fontsize=10, title='ManMark')
computing neighbors
using 'X_pca' with n_pcs = 12
finished: added to `.uns['neighbors']`
`.obsp['distances']`, distances for each pair of neighbors
`.obsp['connectivities']`, weighted adjacency matrix (0:00:01)
computing UMAP
finished: added
'X_umap', UMAP coordinates (adata.obsm) (0:00:10)

[23]:
sc.pl.umap(new_adata, color=['CellType'], legend_loc=None, frameon=False, size=5., legend_fontsize=10, title='')

[24]:
sc.settings.set_figure_params(dpi=60, facecolor='white')
sc.pl.umap(new_adata, color=selected_genes, color_map="inferno")

[25]:
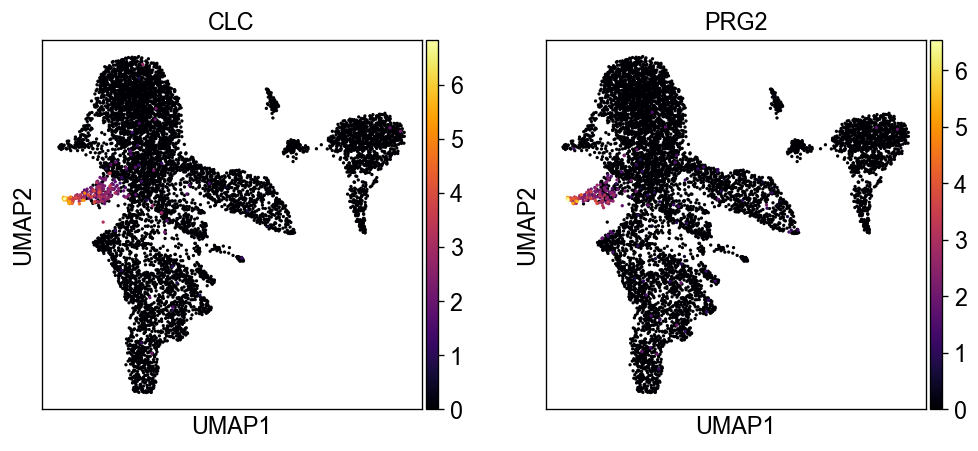
sc.settings.set_figure_params(dpi=60, facecolor='white')
sc.pl.umap(new_adata, color=['CLC', 'PRG2'], color_map='inferno')
[26]:
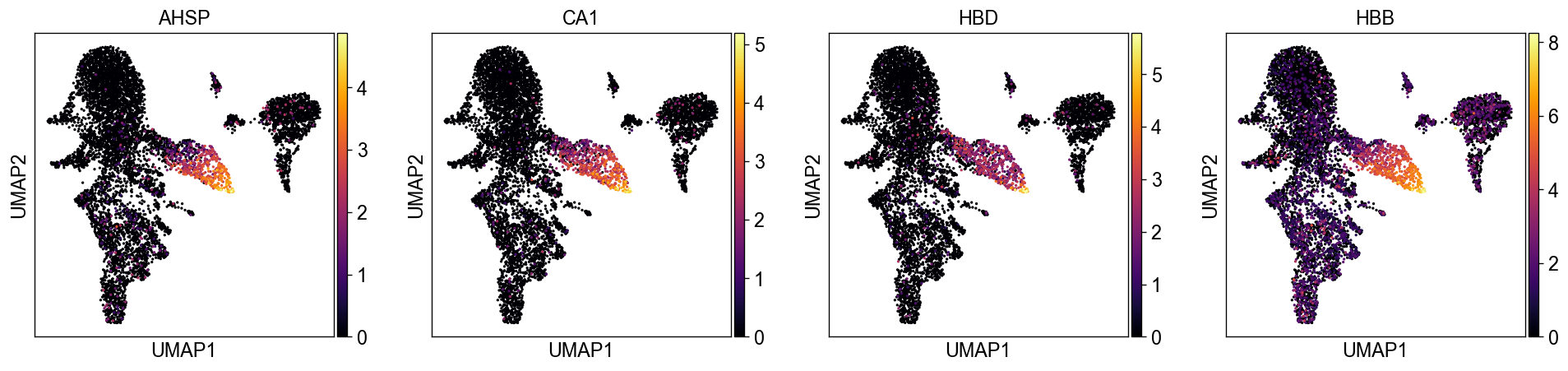
sc.settings.set_figure_params(dpi=60, facecolor='white')
sc.pl.umap(new_adata, color=['AHSP', 'CA1', 'HBD', 'HBB'], color_map='inferno')
[ ]:
[27]:
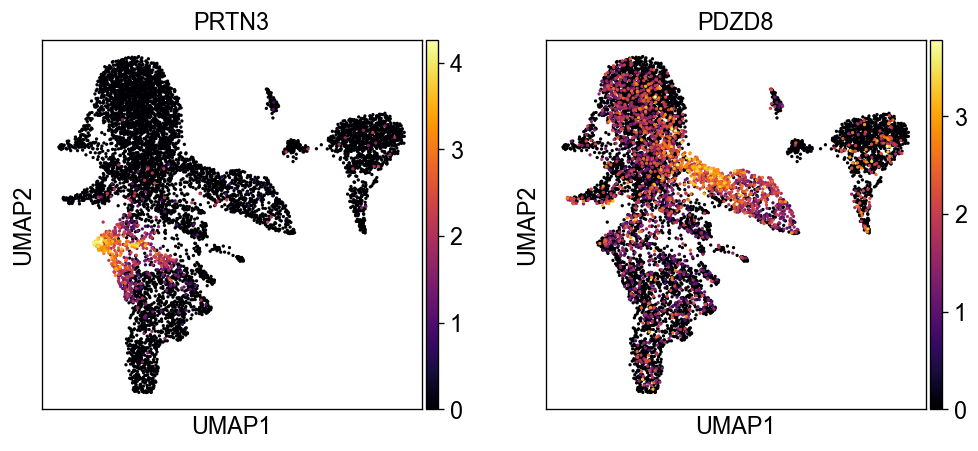
sc.settings.set_figure_params(dpi=60, facecolor='white')
sc.pl.umap(new_adata, color=['PRTN3', 'PDZD8'], color_map='inferno')
[28]:
sc.settings.set_figure_params(dpi=60, facecolor='white')
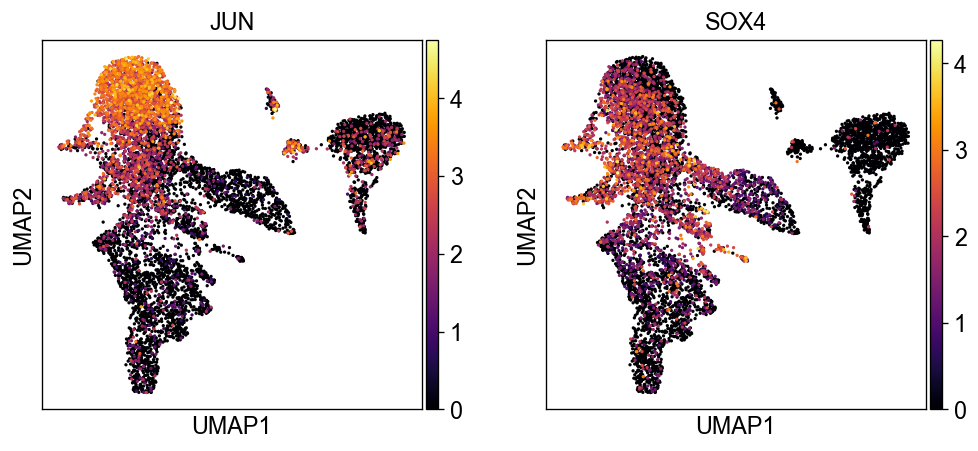
sc.pl.umap(new_adata, color=['JUN', 'SOX4'], color_map='inferno')
[29]:
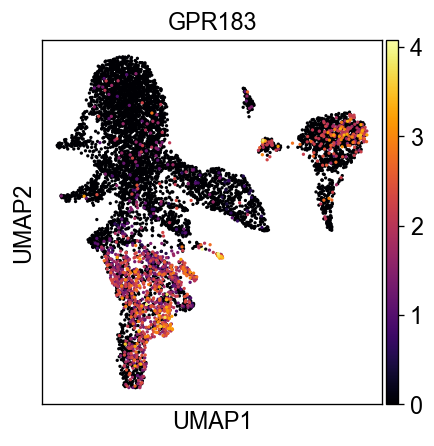
sc.settings.set_figure_params(dpi=60, facecolor='white')
sc.pl.umap(new_adata, color=['GPR183'], color_map='inferno')
[30]:
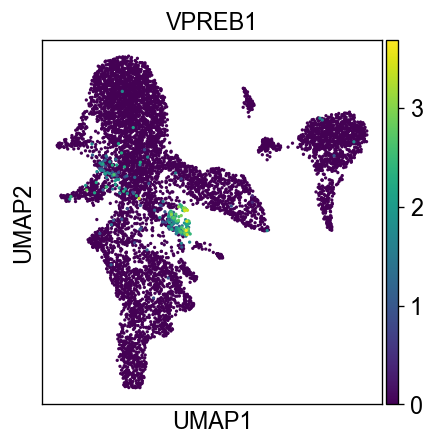
sc.settings.set_figure_params(dpi=60, facecolor='white')
sc.pl.umap(new_adata, color=['VPREB1'])
[31]:
weight_df = pd.DataFrame(model.w[model.w > 0.], index=adata.var_names[model.w > 0.])
weight_df.to_pickle("bm-weights.pkl")
weight_df.to_csv("bm-weigths.csv")