198 Pan-Cancer Cell Lines Marker Selection
[1]:
import scanpy as sc
import os
import pandas as pd
import numpy as np
import pickle as pkl
import matplotlib as mpl
import matplotlib.pyplot as plt
import scipy.stats
from scipy.io import mmread
sc.settings.verbosity = 3
[2]:
obs = pd.read_table("../../pancancer/Metadata.txt", index_col=0, skiprows=[1])
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\IPython\core\interactiveshell.py:3337: DtypeWarning: Columns (2) have mixed types.Specify dtype option on import or set low_memory=False.
if (await self.run_code(code, result, async_=asy)):
[3]:
#adata = sc.read_text("../../pancancer/GSE157220_CPM_data.txt.gz")
#adata = adata.T
[4]:
#adata.write_h5ad("../../pancancer/GSE157220_CPM_data.h5ad")
[5]:
adata = sc.read_h5ad("../../pancancer/GSE157220_CPM_data.h5ad")
[6]:
adata.obs = obs.loc[adata.obs_names]
[7]:
sc.settings.set_figure_params(dpi=80, facecolor='white')
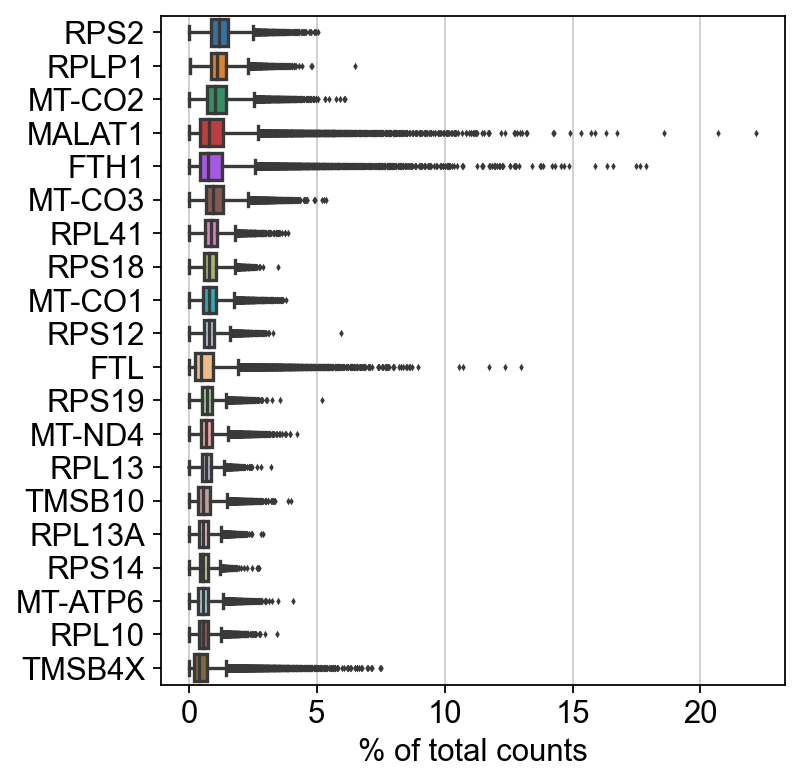
sc.pl.highest_expr_genes(adata, n_top=20)
normalizing counts per cell
finished (0:00:02)
[8]:
#sc.pp.filter_cells(adata, min_genes=100)
#sc.pp.filter_genes(adata, min_cells=5)
[9]:
# adata = adata[adata.obs.pct_counts_mt < 25, :]
[10]:
sc.pp.normalize_total(adata, target_sum=1e4)
normalizing counts per cell
finished (0:00:00)
[11]:
sc.pp.log1p(adata)
[12]:
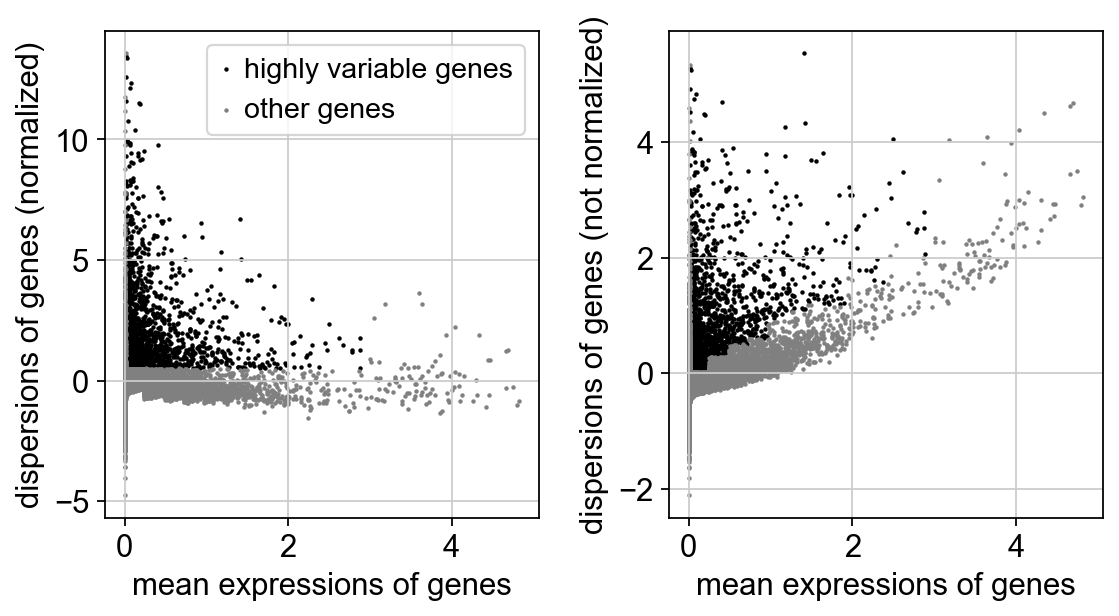
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5)
sc.pl.highly_variable_genes(adata)
extracting highly variable genes
finished (0:00:12)
--> added
'highly_variable', boolean vector (adata.var)
'means', float vector (adata.var)
'dispersions', float vector (adata.var)
'dispersions_norm', float vector (adata.var)
[13]:
adata.var.highly_variable.sum()
[13]:
2208
[14]:
adata.raw = adata
[15]:
adata = adata[:, adata.var.highly_variable]
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1094: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if not is_categorical(df_full[k]):
[16]:
sc.pp.scale(adata, max_value=10)
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\scanpy\preprocessing\_simple.py:806: UserWarning: Revieved a view of an AnnData. Making a copy.
view_to_actual(adata)
[17]:
adata.obs.head()
[17]:
| Cell_line | Pool_ID | Cancer_type | Genes_expressed | Discrete_cluster_minpts5_eps1.8 | Discrete_cluster_minpts5_eps1.5 | Discrete_cluster_minpts5_eps1.2 | CNA_subclone | SkinPig_score | EMTI_score | EMTII_score | EMTIII_score | IFNResp_score | p53Sen_score | EpiSen_score | StressResp_score | ProtMatu_score | ProtDegra_score | G1/S_score | G2/M_score | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AAACCTGAGACATAAC-1-18 | NCIH2126_LUNG | 18 | Lung Cancer | 4318 | NaN | NaN | NaN | NaN | 0.166 | -0.045 | -0.935 | -0.935 | 0.130 | 0.619 | 1.869 | -0.004 | 0.805 | 0.896 | 0.424 | -1.125 |
| AACGTTGTCACCCGAG-1-18 | NCIH2126_LUNG | 18 | Lung Cancer | 5200 | NaN | NaN | NaN | NaN | -0.213 | 0.035 | -1.027 | -1.027 | 0.066 | 1.049 | 1.267 | 0.252 | 1.299 | 1.610 | 0.624 | -0.048 |
| AACTGGTAGACACGAC-1-18 | NCIH2126_LUNG | 18 | Lung Cancer | 4004 | NaN | NaN | NaN | NaN | -0.101 | -0.183 | -0.677 | -0.677 | 0.304 | 0.822 | 2.401 | 0.141 | 0.451 | 1.225 | -0.795 | 0.064 |
| AACTGGTAGGGCTTGA-1-18 | NCIH2126_LUNG | 18 | Lung Cancer | 4295 | NaN | NaN | NaN | NaN | -0.014 | -0.093 | -0.735 | -0.735 | 0.094 | 0.834 | 2.282 | 0.150 | 0.267 | 0.892 | -0.238 | 1.118 |
| AACTGGTAGTACTTGC-1-18 | NCIH2126_LUNG | 18 | Lung Cancer | 4842 | NaN | NaN | NaN | NaN | 0.006 | -0.055 | -0.821 | -0.821 | 0.034 | 0.960 | 1.400 | -0.012 | -0.276 | -0.428 | 0.267 | 0.791 |
[18]:
sc.settings.set_figure_params(dpi=100, facecolor='white')
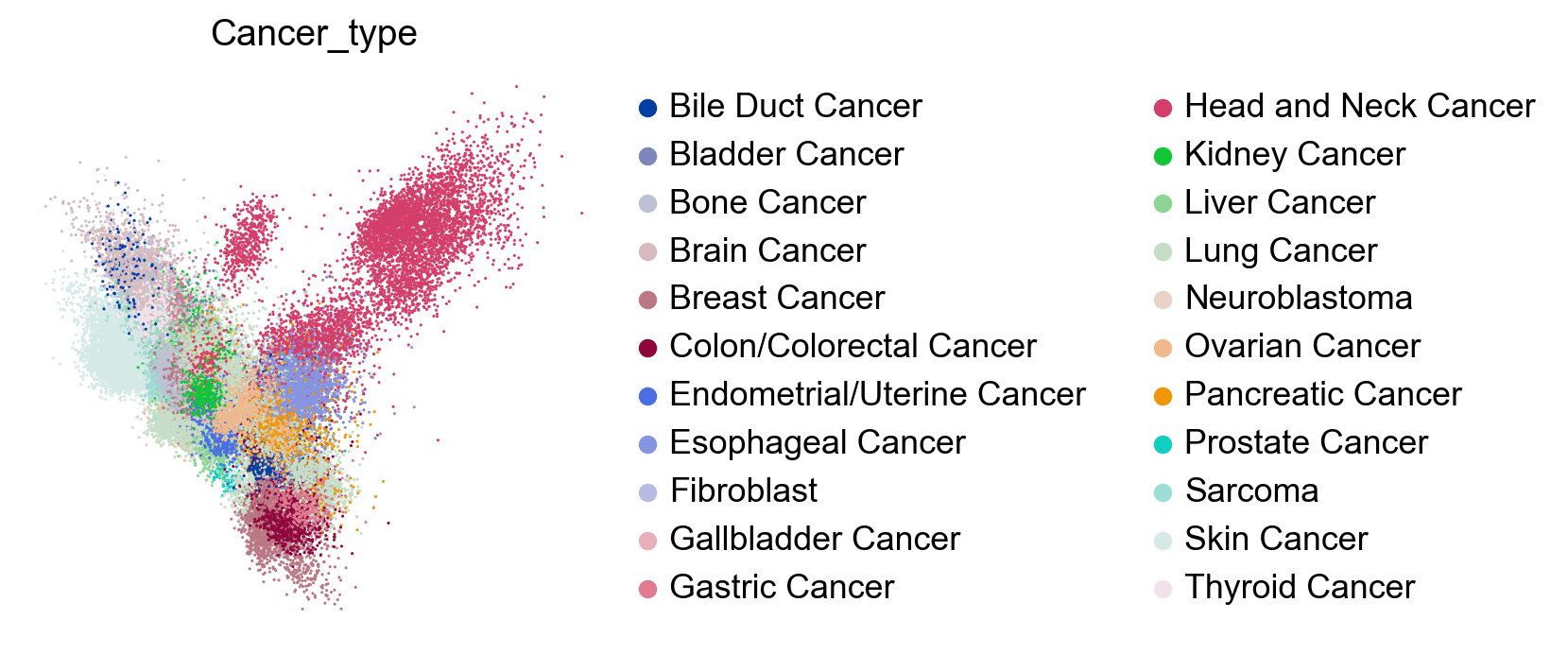
sc.tl.pca(adata, svd_solver='arpack', n_comps=100)
sc.pl.pca(adata, color=['Cancer_type'], size=5., frameon=False)
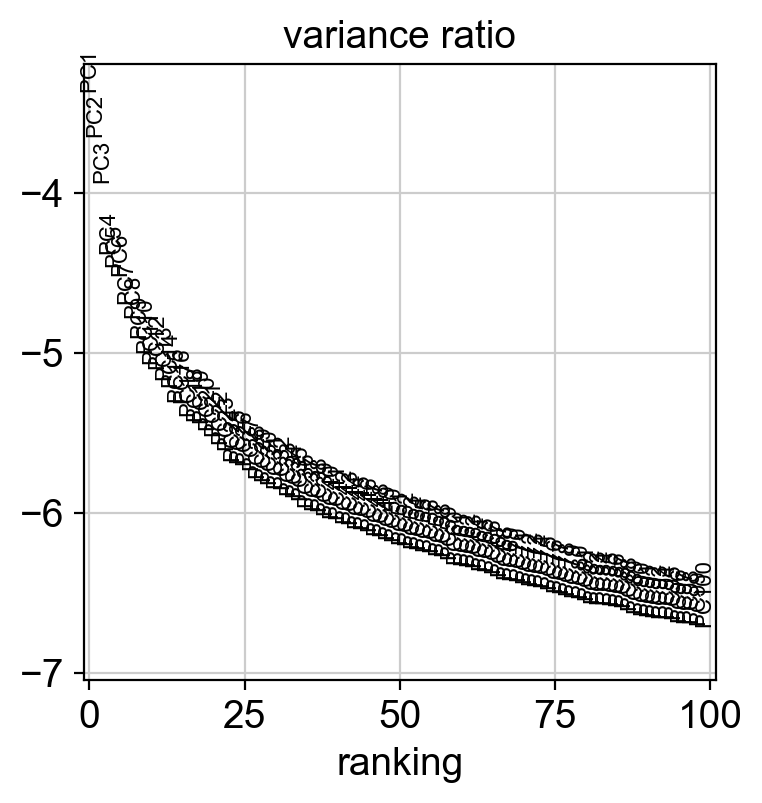
sc.pl.pca_variance_ratio(adata, log=True, n_pcs=100)
computing PCA
on highly variable genes
with n_comps=100
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1094: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if not is_categorical(df_full[k]):
finished (0:00:09)
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1192: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if is_string_dtype(df[key]) and not is_categorical(df[key])
... storing 'Cell_line' as categorical
... storing 'Pool_ID' as categorical
... storing 'Cancer_type' as categorical
... storing 'Discrete_cluster_minpts5_eps1.8' as categorical
... storing 'Discrete_cluster_minpts5_eps1.5' as categorical
... storing 'Discrete_cluster_minpts5_eps1.2' as categorical
... storing 'CNA_subclone' as categorical
[19]:
sc.pp.neighbors(adata, n_pcs=100)
sc.tl.umap(adata)
sc.settings.set_figure_params(dpi=100, facecolor='white')
sc.pl.umap(adata, color=['Cancer_type'], frameon=False, size=5., legend_fontsize=10, title="")
computing neighbors
using 'X_pca' with n_pcs = 100
finished: added to `.uns['neighbors']`
`.obsp['distances']`, distances for each pair of neighbors
`.obsp['connectivities']`, weighted adjacency matrix (0:00:19)
computing UMAP
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\sklearn\manifold\_spectral_embedding.py:236: UserWarning: Graph is not fully connected, spectral embedding may not work as expected.
warnings.warn("Graph is not fully connected, spectral embedding"
finished: added
'X_umap', UMAP coordinates (adata.obsm) (0:00:47)
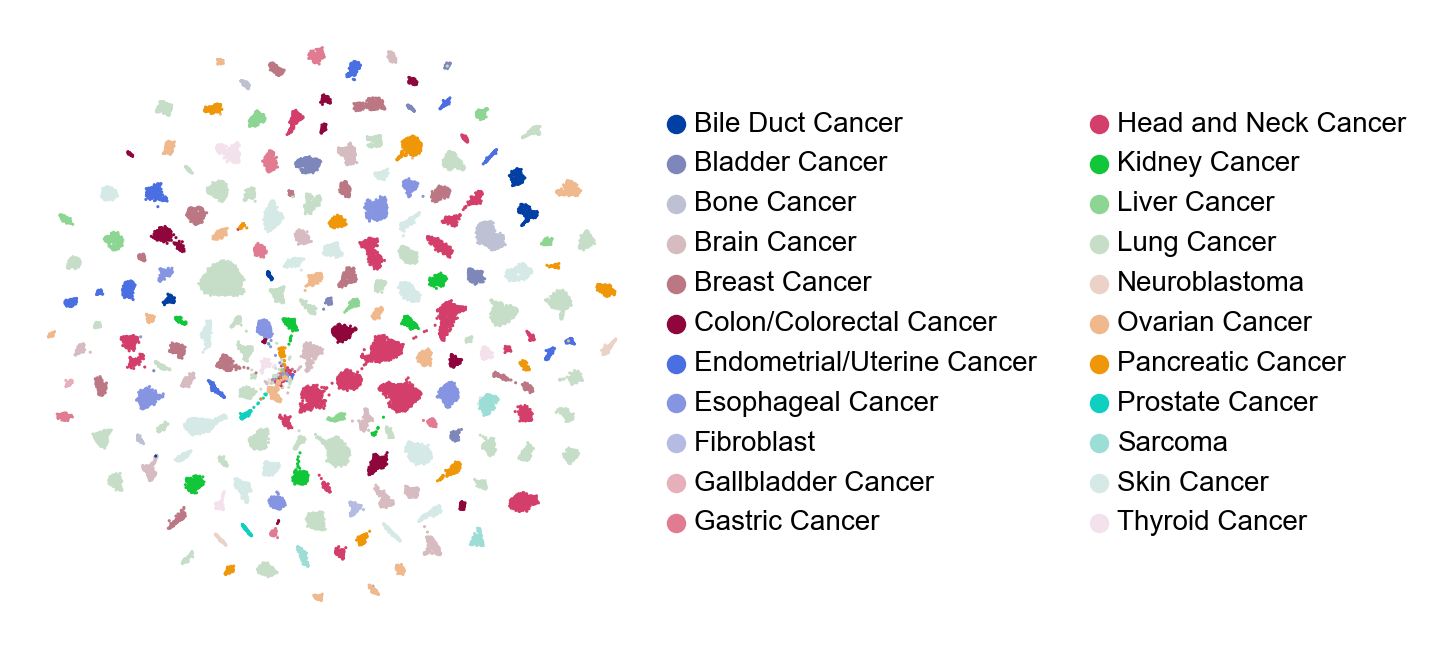
[20]:
adata.obs.Cell_line.unique()
[20]:
['NCIH2126_LUNG', 'SW579_THYROID', 'C32_SKIN', 'NCIH446_LUNG', 'HEC251_ENDOMETRIUM', ..., 'SCC9_UPPER_AERODIGESTIVE_TRACT', 'JHU011_UPPER_AERODIGESTIVE_TRACT', '93VU_UPPER_AERODIGESTIVE_TRACT', 'SCC90_UPPER_AERODIGESTIVE_TRACT', 'JHU006_UPPER_AERODIGESTIVE_TRACT']
Length: 198
Categories (198, object): ['NCIH2126_LUNG', 'SW579_THYROID', 'C32_SKIN', 'NCIH446_LUNG', ..., 'JHU011_UPPER_AERODIGESTIVE_TRACT', '93VU_UPPER_AERODIGESTIVE_TRACT', 'SCC90_UPPER_AERODIGESTIVE_TRACT', 'JHU006_UPPER_AERODIGESTIVE_TRACT']
[21]:
sc.settings.set_figure_params(dpi=300, facecolor='white')
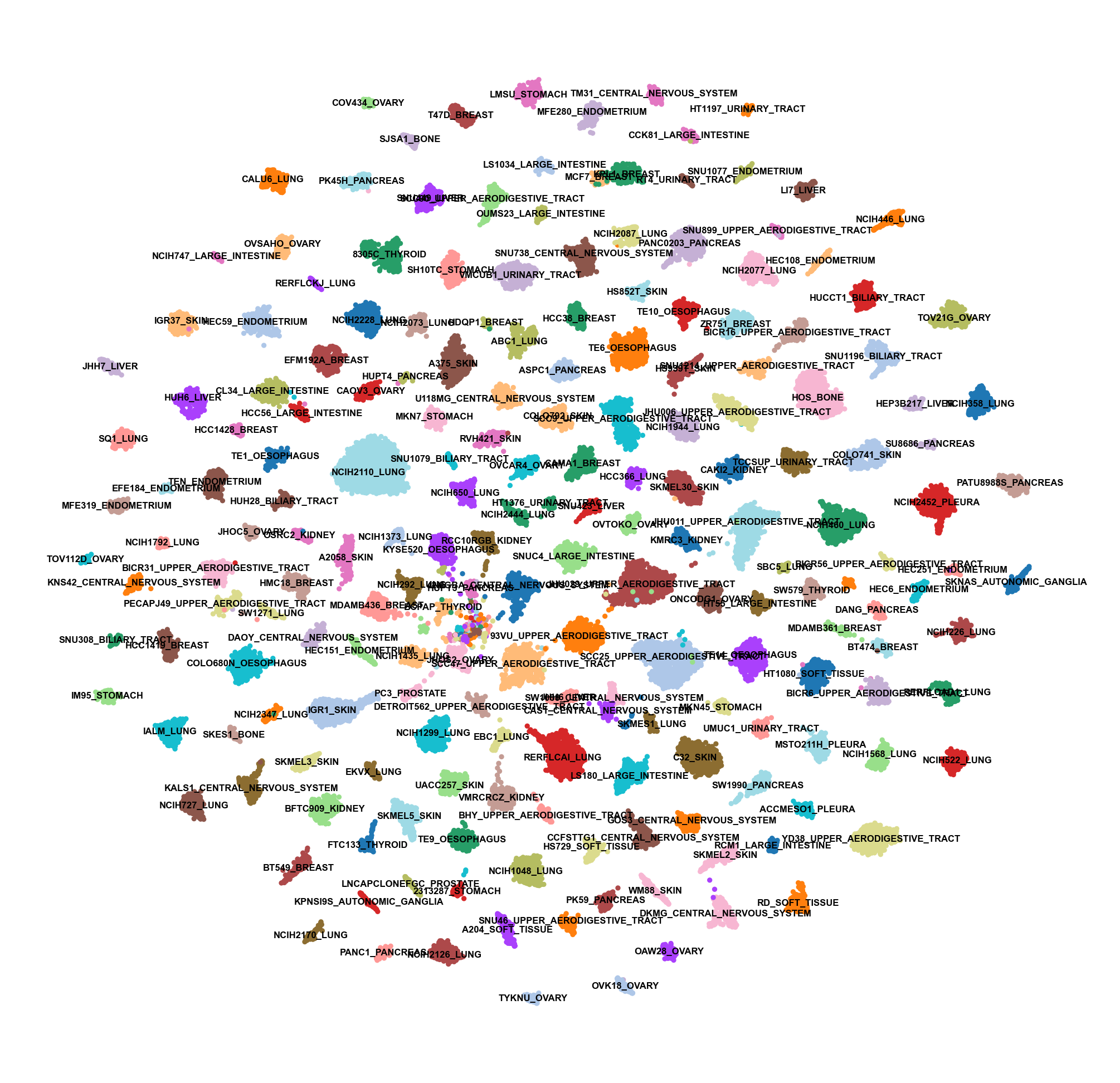
sc.pl.umap(adata, color=['Cell_line'], frameon=False, legend_loc="on data", size=5., legend_fontsize=2., title="", palette=sc.pl.palettes.default_20)
WARNING: Length of palette colors is smaller than the number of categories (palette length: 20, categories length: 198. Some categories will have the same color.
SCMER Feature Selection
[22]:
import sys
sys.path.insert(0,'..')
import scmer
model = scmer.UmapL1(w=1., lasso=2e-4, ridge=2e-4, n_pcs=50, perplexity=100., use_beta_in_Q=True, n_threads=6, max_outer_iter=2)
model.fit(adata.X, batches=adata.obs['Cancer_type'].values)
Batch Bile Duct Cancer with 739 instances.
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 1.378048
Done. Elapsed time: 2.52 seconds. Total: 2.52 seconds.
Batch Bladder Cancer with 1291 instances.
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 1.198487
Done. Elapsed time: 3.19 seconds. Total: 5.71 seconds.
Batch Bone Cancer with 1030 instances.
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 1.069480
Done. Elapsed time: 2.78 seconds. Total: 8.49 seconds.
Batch Brain Cancer with 2967 instances.
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 1.197244
Done. Elapsed time: 8.79 seconds. Total: 17.28 seconds.
Batch Breast Cancer with 3285 instances.
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 1.118649
Done. Elapsed time: 9.58 seconds. Total: 26.86 seconds.
Batch Colon/Colorectal Cancer with 2104 instances.
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 1.228417
Done. Elapsed time: 5.02 seconds. Total: 31.88 seconds.
Batch Endometrial/Uterine Cancer with 2057 instances.
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 1.109343
Done. Elapsed time: 4.91 seconds. Total: 36.79 seconds.
Batch Esophageal Cancer with 2544 instances.
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 1.095491
Done. Elapsed time: 6.43 seconds. Total: 43.21 seconds.
Batch Fibroblast with 215 instances.
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 1.633770
Done. Elapsed time: 1.75 seconds. Total: 44.96 seconds.
Batch Gallbladder Cancer with 94 instances.
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 1.644982
Done. Elapsed time: 1.71 seconds. Total: 46.67 seconds.
Batch Gastric Cancer with 1270 instances.
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 1.206267
Done. Elapsed time: 2.95 seconds. Total: 49.62 seconds.
Batch Head and Neck Cancer with 7102 instances.
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 1.041096
Done. Elapsed time: 37.56 seconds. Total: 87.18 seconds.
Batch Kidney Cancer with 1561 instances.
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 1.204537
Done. Elapsed time: 3.46 seconds. Total: 90.64 seconds.
Batch Liver Cancer with 1550 instances.
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 1.269934
Done. Elapsed time: 3.62 seconds. Total: 94.26 seconds.
Batch Lung Cancer with 12842 instances.
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 0.886191
Done. Elapsed time: 116.71 seconds. Total: 210.97 seconds.
Batch Neuroblastoma with 355 instances.
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 1.399799
Done. Elapsed time: 1.83 seconds. Total: 212.80 seconds.
Batch Ovarian Cancer with 2495 instances.
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 1.187534
Done. Elapsed time: 6.15 seconds. Total: 218.95 seconds.
Batch Pancreatic Cancer with 2368 instances.
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 1.146014
Done. Elapsed time: 5.84 seconds. Total: 224.79 seconds.
Batch Prostate Cancer with 298 instances.
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 1.508120
Done. Elapsed time: 1.84 seconds. Total: 226.63 seconds.
Batch Sarcoma with 1027 instances.
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 1.249106
Done. Elapsed time: 2.69 seconds. Total: 229.32 seconds.
Batch Skin Cancer with 5351 instances.
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 1.135544
Done. Elapsed time: 22.14 seconds. Total: 251.46 seconds.
Batch Thyroid Cancer with 968 instances.
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 1.312984
Done. Elapsed time: 2.46 seconds. Total: 253.91 seconds.
Creating model without batches...
Optimizing using OWLQN (because lasso is nonzero)...
0 loss (before this step): 3.1993868350982666 Nonzero (after): 251 Elapsed time: 833.58 seconds. Total: 1087.50 seconds.
1 loss (before this step): 2.42031192779541 Nonzero (after): 247 Elapsed time: 963.12 seconds. Total: 2050.61 seconds.
Final loss: 2.36572003364563 Nonzero: 247 Elapsed time: 13.43 seconds. Total: 2064.05 seconds.
[22]:
<scmer._umap_l1.UmapL1 at 0x2151eb28208>
Validation
[29]:
rhp_df = pd.read_excel('../../pancancer/41588_2020_726_MOESM3_ESM.xlsx', sheet_name='Table S4', skiprows=3)
rhp_nmf = {rhp_df.columns[i]: rhp_df[rhp_df.columns[i]].dropna().tolist() for i in range(12)}
[30]:
rhp_df = pd.read_excel('../../pancancer/41588_2020_726_MOESM3_ESM.xlsx', sheet_name='Table S7', skiprows=3)
rhp_vivo = {rhp_df.columns[i]: rhp_df[rhp_df.columns[i]].dropna().tolist() for i in range(21)}
[25]:
from IPython.display import Markdown, display
def printmd(string):
display(Markdown(string))
established_markers = rhp_nmf
selected_markers = adata.var_names[model.w > 0]
all_markers = adata.var_names.tolist()
for i in established_markers:
temp = " ".join((i, str(len(set(established_markers[i]).intersection(set(selected_markers)))),
'/', str(len(set(established_markers[i]).intersection(set(all_markers)))),
"(", str(len(established_markers[i])), "): "))
printmd(temp + '_' + '_, _'.join(set(established_markers[i]).intersection(set(selected_markers))) + '_')
Cell Cycle - G1/S 2 / 12 ( 46 ): HIST1H1C, HMGB2
Cell Cycle - G2/M 7 / 30 ( 62 ): CENPF, TOP2A, AURKA, CKS2, UBE2S, MKI67, HMGB2
Skin Pigmentation 4 / 37 ( 47 ): CAPN3, APOE, MLANA, DCT
EMT I 23 / 63 ( 79 ): CAV1, CXCL1, LIMCH1, CCND1, FN1, C12orf75, DKK1, PEG10, SFRP1, KRT7, S100A16, THBS1, ACTG2, S100A4, MYL9, FOSL1, DCBLD2, KRT81, PRSS23, PMEPA1, TGFBI, TPM1, TGM2
EMT II 18 / 24 ( 28 ): VIM, AXL, TAGLN, FN1, IL32, CST6, INHBA, FST, PMEPA1, THBS1, SERPINE1, TPM1, AKAP12, IGFBP7, MYL9, IGFBP3, LAMC2, PRSS23
IFN Response 5 / 39 ( 49 ): IFIT3, ISG15, HLA-B, IL7R, OASL
EMT III 15 / 43 ( 49 ): NEAT1, SLPI, FN1, IL32, G0S2, INHBA, CST6, LAMB3, S100A14, SERPINE1, WFDC2, DMKN, LCN2, LAMC2, KLK10
p53-Dependent Senescence 6 / 13 ( 19 ): S100P, ISG15, KRT19, NEAT1, SLPI, IFI27
Epithelial Senescence 11 / 37 ( 38 ): S100P, NEAT1, SLPI, S100A14, CXCL1, LY6D, ADIRF, AGR2, WFDC2, KRT13, LCN2
Stress Response 0 / 29 ( 36 ): __
Protein Maturation 1 / 13 ( 34 ): LAMB3
Proteasomal Degradation 2 / 8 ( 46 ): PRDX1, EIF4A1
[26]:
established_markers = rhp_vivo
selected_markers = adata.var_names[model.w > 0]
all_markers = adata.var_names.tolist()
for i in established_markers:
temp = " ".join((i, str(len(set(established_markers[i]).intersection(set(selected_markers)))),
'/', str(len(set(established_markers[i]).intersection(set(all_markers)))),
"(", str(len(established_markers[i])), "): "))
printmd(temp + '_' + '_, _'.join(set(established_markers[i]).intersection(set(selected_markers))) + '_')
GBM.MES2_orig 4 / 32 ( 50 ): SLC2A3, IGFBP3, VIM, AKAP12
GBM.MES1_orig 14 / 35 ( 50 ): S100A11, VIM, IFITM3, MGST1, FN1, S100A16, APOE, SERPINE1, S100A10, SPP1, IGFBP7, MT1E, MGP, TAGLN2
GBM.AC_orig 2 / 18 ( 39 ): S100A16, RAMP1
GBM.OPC_orig 2 / 18 ( 50 ): FABP5, THY1
GBM.NPC1_orig 2 / 19 ( 50 ): SOX11, BEX1
GBM.NPC2_orig 2 / 26 ( 50 ): SOX11, UCHL1
HNSCC.PEMT_orig 20 / 62 ( 100 ): CAV1, TAGLN, MMP1, INHBA, IGFBP3, LAMC2, OCIAD2, SERPINE1, ARPC1B, IL32, MMP2, TPM4, LAMB3, COL1A1, THBS1, IGFBP7, PRSS23, VIM, TGFBI, TPM1
HNSCC.Epidif.1_orig 10 / 62 ( 100 ): S100P, SLPI, CEACAM6, ALDH3B2, LY6D, HIST1H1C, DMKN, PDZK1IP1, LCN2, KLK10
HNSCC.Epidif.2_orig 6 / 34 ( 100 ): AKR1B10, S100A14, FABP5, S100A16, LY6D, MAL2
HNSCC.Stress_orig 4 / 63 ( 100 ): ID1, CD74, LAMB3, FOSL1
HNSCC.Hypoxia_orig 6 / 47 ( 100 ): IGFBP2, SERPINE1, GJB6, HIST1H1C, IGFBP3, ENO1
melanoma.MITF_orig 4 / 43 ( 100 ): CAPN3, ARPC1B, APOE, MLANA
melanoma.AXL_orig 11 / 53 ( 100 ): AXL, FN1, S100A16, SERPINE1, S100A10, SH3BGRL3, S100A4, FOSL1, IGFBP3, LCN2, SLC16A3
melanoma.Inflammatory_orig 8 / 68 ( 229 ): CKS2, SLC2A3, TM4SF1, AKR1B1, PAGE5, SERPINE1, SPP1, SLC16A3
pemt.metaprogram 14 / 39 ( 45 ): VIM, TAGLN, MMP1, KRT8, INHBA, IL32, MMP2, LAMB3, THBS1, SERPINE1, TGFBI, TPM1, IGFBP7, LAMC2
HNSCC.episen_curr 16 / 61 ( 85 ): S100P, AKR1B10, ISG15, SLPI, CEACAM6, FABP5, CST6, IGFL2, ALDH3B2, LY6D, CALML5, DMKN, KRT23, PDZK1IP1, LCN2, KLK10
hypoxia_curr 2 / 23 ( 40 ): IGFBP3, SLC2A3
immune.resp_curr 8 / 32 ( 46 ): CD74, RARRES2, IFI27, COL1A1, THY1, S100A4, COL1A2, IFITM2
HNSCC.melanoma.stress_curr 5 / 46 ( 63 ): CKS2, IL32, CXCL1, ID1, FOSL1
g1.s_curr 1 / 11 ( 55 ): HMGB2
g2.m_curr 5 / 25 ( 69 ): CENPF, TOP2A, AURKA, CKS2, HMGB2
[40]:
pd.DataFrame({k: ", ".join(v) for k, v in rhp_nmf.items()}, index=['Genes']).T.to_csv(
"dump-data/Gene-sets/Pancancer-RHP-NMF.csv")
pd.DataFrame({k: ", ".join([str(i) for i in v]) for k, v in rhp_vivo.items()}, index=['Genes']).T.to_csv(
"dump-data/Gene-sets/Pancancer-RHP-Vivo.csv")
[41]:
dump_data = pd.DataFrame(index=adata.obs_names)
dump_data[['Cell_line']] = adata.obs[['Cell_line']]
dump_data[['Original_UMAP1', 'Original_UMAP2']] = adata.obsm['X_umap']
dump_features = list(set(['PRDX1', 'CDC20']))
dump_data[dump_features] = adata.raw.to_adata()[:, dump_features].X
dump_data
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1094: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if not is_categorical(df_full[k]):
[41]:
| Cell_line | Original_UMAP1 | Original_UMAP2 | PRDX1 | CDC20 | |
|---|---|---|---|---|---|
| AAACCTGAGACATAAC-1-18 | NCIH2126_LUNG | -4.693660 | -10.419457 | 3.149034 | 0.000000 |
| AACGTTGTCACCCGAG-1-18 | NCIH2126_LUNG | -4.332566 | -10.322994 | 3.330684 | 1.079388 |
| AACTGGTAGACACGAC-1-18 | NCIH2126_LUNG | -4.886784 | -10.156097 | 3.130302 | 1.681977 |
| AACTGGTAGGGCTTGA-1-18 | NCIH2126_LUNG | -4.574833 | -9.963341 | 3.531188 | 1.848316 |
| AACTGGTAGTACTTGC-1-18 | NCIH2126_LUNG | -4.277936 | -10.106932 | 1.808250 | 1.068981 |
| ... | ... | ... | ... | ... | ... |
| c4722 | JHU006_UPPER_AERODIGESTIVE_TRACT | 6.447311 | 11.874496 | 2.580448 | 0.000000 |
| c4724 | JHU006_UPPER_AERODIGESTIVE_TRACT | 6.758837 | 11.946816 | 2.867576 | 1.189283 |
| c4731 | JHU006_UPPER_AERODIGESTIVE_TRACT | 6.952734 | 11.815464 | 2.529585 | 1.426004 |
| c4735 | JHU006_UPPER_AERODIGESTIVE_TRACT | 7.577816 | 11.011492 | 2.643856 | 0.626556 |
| c4741 | JHU006_UPPER_AERODIGESTIVE_TRACT | 7.468618 | 11.110316 | 3.266989 | 0.244731 |
53513 rows × 5 columns
[42]:
dump_data.to_csv("dump-data/Fig-S3.csv")
[27]:
weight_df = pd.DataFrame(model.w[model.w > 0.], index=adata.var_names[model.w > 0.])
weight_df.to_pickle("dump-data/pancancer-weights.pkl")
weight_df.to_csv("dump-data/pancancer-weigths.csv")