Dexamethasone Treated A549 Lung Cancer Cell Line (Cao et al.)
Data: GSE117089
Article Joint profiling of chromatin accessibility and gene expression in thousands of single cells
[1]:
import scanpy as sc
import os
import pandas as pd
import numpy as np
import pickle as pkl
import matplotlib as mpl
import matplotlib.pyplot as plt
import scipy.stats
import seaborn as sns
import pickle as pkl
from scipy.io import mmread
sc.settings.verbosity = 3
[2]:
import sys
sys.path.insert(0,'..')
import scmer
Data Preparation
[3]:
def read_data(path):
adata = sc.read_mtx(path + "_gene_count.txt.gz").T
adata.obs = pd.read_csv(path + "_cell.txt.gz", index_col=0, compression='gzip')
adata.var = pd.read_csv(path + "_gene.txt.gz", index_col=0, compression='gzip')
return adata
coassay_adata = read_data("../../sci-car/GSM3271040_RNA_sciCAR_A549")
coassay_adata = coassay_adata[coassay_adata.obs.cell_name == 'A549', coassay_adata.var.index.str.contains('^ENSG')]
rna_only_adata = read_data("../../sci-car/GSM3271042_RNA_only_A549")
rna_only_adata = rna_only_adata[:, rna_only_adata.var.index.str.contains('^ENSG')]
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1094: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if not is_categorical(df_full[k]):
[4]:
print(coassay_adata)
print(rna_only_adata)
View of AnnData object with n_obs × n_vars = 4277 × 63568
obs: 'cell_name', 'experiment', 'treatment_time'
var: 'gene_type', 'gene_short_name'
View of AnnData object with n_obs × n_vars = 1873 × 63568
obs: 'cell_name', 'experiment', 'treatment_time'
var: 'gene_type', 'gene_short_name'
[5]:
adata = sc.concat(adatas=[coassay_adata, rna_only_adata])
adata
[5]:
AnnData object with n_obs × n_vars = 6150 × 63568
obs: 'cell_name', 'experiment', 'treatment_time'
[6]:
adata.obs['treatment_time'] = adata.obs['treatment_time'].astype(int).astype(str)
adata.obs
[6]:
| cell_name | experiment | treatment_time | |
|---|---|---|---|
| sample | |||
| sci-RNA-A-001.AAGTACGTTA | A549 | coassay | 3 |
| sci-RNA-A-001.TCTCTCATCC | A549 | coassay | 0 |
| sci-RNA-A-001.TCCGCCGGTC | A549 | coassay | 3 |
| sci-RNA-A-001.TTCTATAGAG | A549 | coassay | 1 |
| sci-RNA-A-001.CGTCTATGAA | A549 | coassay | 1 |
| ... | ... | ... | ... |
| co-RNA-only-192.TTCGCTGCCT | A549 | RNA_only | 1 |
| co-RNA-only-192.TTCTCTACTA | A549 | RNA_only | 1 |
| co-RNA-only-192.TTGCAGCATT | A549 | RNA_only | 1 |
| co-RNA-only-192.TCCTCTCCGT | A549 | RNA_only | 3 |
| co-RNA-only-192.GGACGACGCA | A549 | RNA_only | 1 |
6150 rows × 3 columns
[7]:
adata.var['gene_type'] = coassay_adata.var.gene_type
adata.var['gene_short_name'] = coassay_adata.var.gene_short_name
[8]:
adata.var.gene_type.value_counts().to_frame("Counts")
[8]:
| Counts | |
|---|---|
| protein_coding | 22705 |
| pseudogene | 15588 |
| lincRNA | 7335 |
| antisense | 5478 |
| miRNA | 3367 |
| misc_RNA | 2175 |
| snRNA | 2067 |
| snoRNA | 1549 |
| processed_transcript | 814 |
| sense_intronic | 767 |
| rRNA | 567 |
| IG_V_pseudogene | 255 |
| sense_overlapping | 208 |
| IG_V_gene | 178 |
| TR_V_gene | 150 |
| TR_J_gene | 82 |
| IG_D_gene | 64 |
| polymorphic_pseudogene | 53 |
| TR_V_pseudogene | 40 |
| IG_J_gene | 24 |
| 3prime_overlapping_ncrna | 24 |
| IG_C_gene | 23 |
| Mt_tRNA | 22 |
| IG_C_pseudogene | 11 |
| IG_J_pseudogene | 6 |
| TR_C_gene | 6 |
| TR_J_pseudogene | 4 |
| TR_D_gene | 3 |
| Mt_rRNA | 2 |
| processed_pseudogene | 1 |
[9]:
def make_unique(x):
d = {}
r = []
for i in x:
if i in d:
d[i] += 1
r.append(i + '__' + str(d[i]))
# print(i, '->', r[-1], sep='\t')
else:
d[i] = 1
r.append(i)
return r
adata.var['gene'] = make_unique(adata.var.gene_short_name)
[10]:
adata.var
[10]:
| gene_type | gene_short_name | gene | |
|---|---|---|---|
| gene_id | |||
| ENSG00000223972.4 | pseudogene | DDX11L1 | DDX11L1 |
| ENSG00000227232.4 | pseudogene | WASH7P | WASH7P |
| ENSG00000243485.2 | lincRNA | MIR1302-11 | MIR1302-11 |
| ENSG00000237613.2 | lincRNA | FAM138A | FAM138A |
| ENSG00000268020.2 | pseudogene | OR4G4P | OR4G4P |
| ... | ... | ... | ... |
| ENSG00000241559.2 | miRNA | CU459202.2 | CU459202.2 |
| ENSG00000264728.1 | miRNA | CU442762.3 | CU442762.3 |
| ENSG00000238667.1 | miRNA | CU442762.2 | CU442762.2 |
| ENSG00000238477.1 | miRNA | CU442762.1 | CU442762.1 |
| ENSG00000271726.1 | miRNA | CU442762.4 | CU442762.4 |
63568 rows × 3 columns
[11]:
adata.var['gene_id'] = adata.var.index
adata.var.index = adata.var['gene']
SCANPY Analysis
[12]:
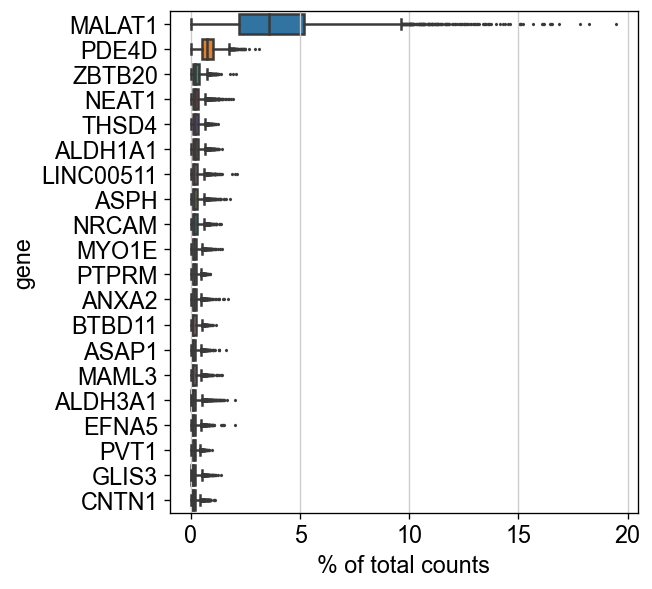
sc.settings.set_figure_params(dpi=60, facecolor='white')
sc.pl.highest_expr_genes(adata, n_top=20)
normalizing counts per cell
finished (0:00:00)
[13]:
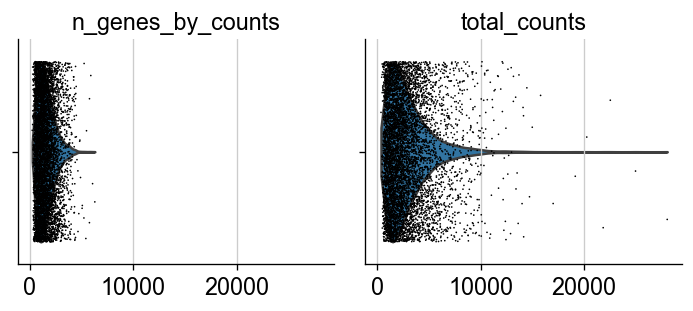
sc.pp.calculate_qc_metrics(adata, percent_top=None, log1p=False, inplace=True)
sc.pl.violin(adata, ['n_genes_by_counts', 'total_counts'], jitter=0.4, multi_panel=True)
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1192: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if is_string_dtype(df[key]) and not is_categorical(df[key])
... storing 'cell_name' as categorical
... storing 'experiment' as categorical
... storing 'treatment_time' as categorical
... storing 'gene_type' as categorical
... storing 'gene_short_name' as categorical
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\seaborn\_core.py:1303: UserWarning: Vertical orientation ignored with only `x` specified.
warnings.warn(single_var_warning.format("Vertical", "x"))
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\seaborn\_core.py:1303: UserWarning: Vertical orientation ignored with only `x` specified.
warnings.warn(single_var_warning.format("Vertical", "x"))
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\seaborn\_core.py:1303: UserWarning: Vertical orientation ignored with only `x` specified.
warnings.warn(single_var_warning.format("Vertical", "x"))
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\seaborn\_core.py:1303: UserWarning: Vertical orientation ignored with only `x` specified.
warnings.warn(single_var_warning.format("Vertical", "x"))
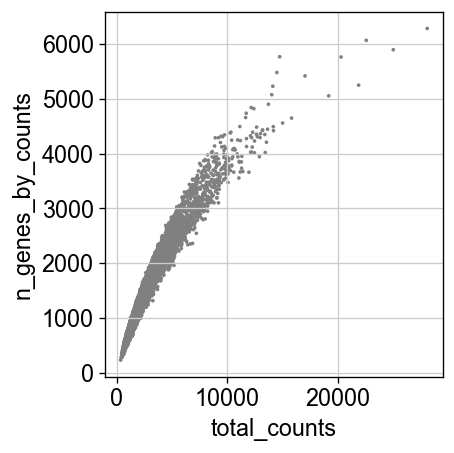
[14]:
sc.pl.scatter(adata, x='total_counts', y='n_genes_by_counts')
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1192: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if is_string_dtype(df[key]) and not is_categorical(df[key])
[15]:
adata = adata[(adata.obs.total_counts >= 500) & (adata.obs.total_counts <= 9100), :]
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1094: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if not is_categorical(df_full[k]):
[16]:
# adata = adata[:, adata.var.gene_type == "protein_coding"]
sc.pp.filter_cells(adata, min_genes=200)
sc.pp.filter_genes(adata, min_cells=3)
Trying to set attribute `.obs` of view, copying.
filtered out 34379 genes that are detected in less than 3 cells
[17]:
sc.pp.normalize_total(adata, target_sum=1e4)
sc.pp.log1p(adata)
normalizing counts per cell
finished (0:00:00)
[18]:
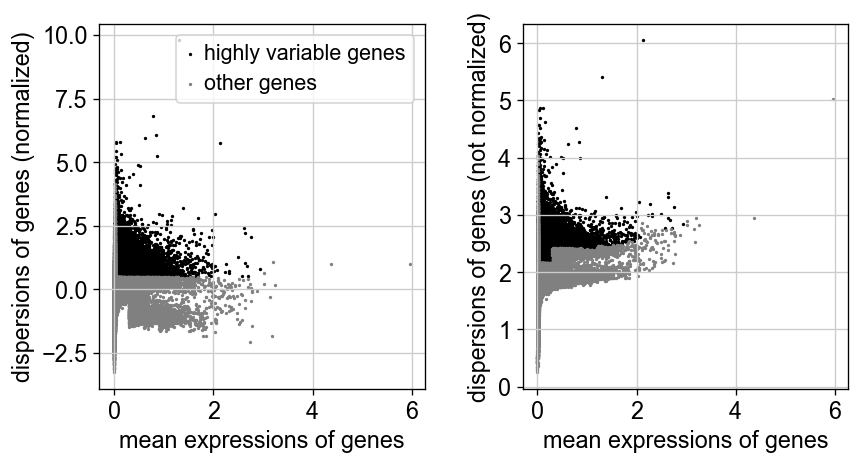
sc.pp.highly_variable_genes(adata)
extracting highly variable genes
finished (0:00:00)
--> added
'highly_variable', boolean vector (adata.var)
'means', float vector (adata.var)
'dispersions', float vector (adata.var)
'dispersions_norm', float vector (adata.var)
[19]:
sc.pl.highly_variable_genes(adata)
[20]:
adata.raw = adata
[21]:
adata = adata[:, adata.var.highly_variable]
[22]:
sc.pp.regress_out(adata, ['total_counts'])
regressing out ['total_counts']
sparse input is densified and may lead to high memory use
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1192: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if is_string_dtype(df[key]) and not is_categorical(df[key])
finished (0:00:29)
[23]:
sc.pp.scale(adata, max_value=10)
adata
[23]:
AnnData object with n_obs × n_vars = 6012 × 6850
obs: 'cell_name', 'experiment', 'treatment_time', 'n_genes_by_counts', 'total_counts', 'n_genes'
var: 'gene_type', 'gene_short_name', 'gene', 'gene_id', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'n_cells', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'mean', 'std'
uns: 'log1p', 'hvg'
[24]:
sc.tl.pca(adata, svd_solver='arpack')
computing PCA
on highly variable genes
with n_comps=50
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1094: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if not is_categorical(df_full[k]):
finished (0:00:04)
[25]:
sc.pl.pca(adata, color=['treatment_time'])
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1192: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if is_string_dtype(df[key]) and not is_categorical(df[key])
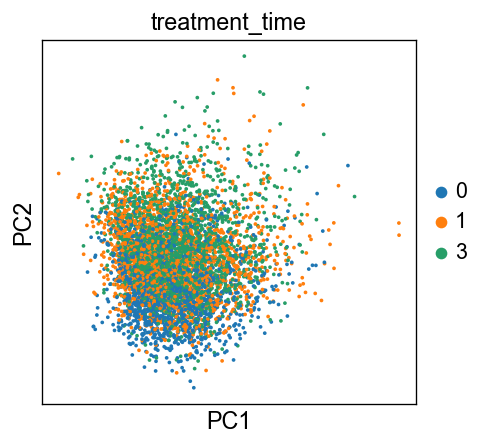
[26]:
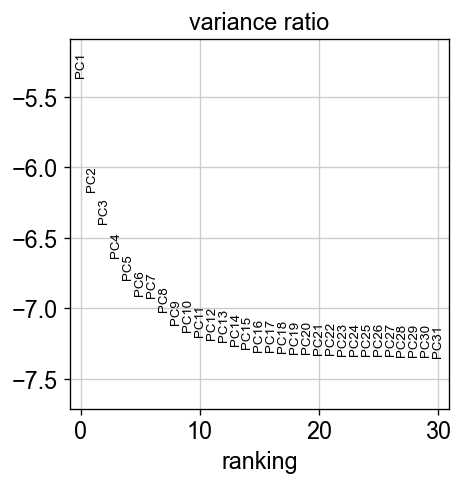
sc.pl.pca_variance_ratio(adata, log=True)
[27]:
sc.pp.neighbors(adata, n_pcs=25)
sc.tl.umap(adata)
df = pd.DataFrame(adata.obsm['X_umap'], columns=['UMAP1', 'UMAP2'])
df['treatment_time'] = [i + '-hour' for i in adata.obs['treatment_time'].tolist()]
sns.jointplot(data=df.sort_values(by='treatment_time', ascending=False), x="UMAP1", y="UMAP2", hue="treatment_time", s=4,
palette=[sns.color_palette("tab10")[i] for i in [2, 0, 1]])
computing neighbors
using 'X_pca' with n_pcs = 25
finished: added to `.uns['neighbors']`
`.obsp['distances']`, distances for each pair of neighbors
`.obsp['connectivities']`, weighted adjacency matrix (0:00:03)
computing UMAP
finished: added
'X_umap', UMAP coordinates (adata.obsm) (0:00:08)
[27]:
<seaborn.axisgrid.JointGrid at 0x21b033325c8>

SCMER Feature Selection
[28]:
model = scmer.UmapL1(w=1., lasso=1e-4, ridge=1e-4, n_pcs=25, perplexity=100., use_beta_in_Q=True, n_threads=6,
max_outer_iter=2)
model.fit(adata.X)
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 0.753229
Done. Elapsed time: 28.75 seconds. Total: 28.75 seconds.
Creating model without batches...
Optimizing using OWLQN (because lasso is nonzero)...
0 loss: 5.043517112731934 Nonzero: 80 Elapsed time: 172.02 seconds. Total: 200.76 seconds.
1 loss: 3.550537586212158 Nonzero: 80 Elapsed time: 178.25 seconds. Total: 379.01 seconds.
final loss: 3.5436885356903076 Nonzero: 80 Elapsed time: 2.73 seconds. Total: 381.74 seconds.
[28]:
<scmer._umap_l1.UmapL1 at 0x21b03c36508>
Validation
PCA and UMAP
[29]:
new_adata = model.transform(adata)
new_adata
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1094: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if not is_categorical(df_full[k]):
[29]:
View of AnnData object with n_obs × n_vars = 6012 × 80
obs: 'cell_name', 'experiment', 'treatment_time', 'n_genes_by_counts', 'total_counts', 'n_genes'
var: 'gene_type', 'gene_short_name', 'gene', 'gene_id', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'n_cells', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'mean', 'std'
uns: 'log1p', 'hvg', 'pca', 'treatment_time_colors', 'neighbors', 'umap'
obsm: 'X_pca', 'X_umap'
varm: 'PCs'
obsp: 'distances', 'connectivities'
[30]:
sc.tl.pca(new_adata, svd_solver='arpack')
sc.pl.pca(new_adata, color=['treatment_time'])
sc.pl.pca_variance_ratio(adata, log=True)
computing PCA
on highly variable genes
with n_comps=50
finished (0:00:00)
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1192: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if is_string_dtype(df[key]) and not is_categorical(df[key])
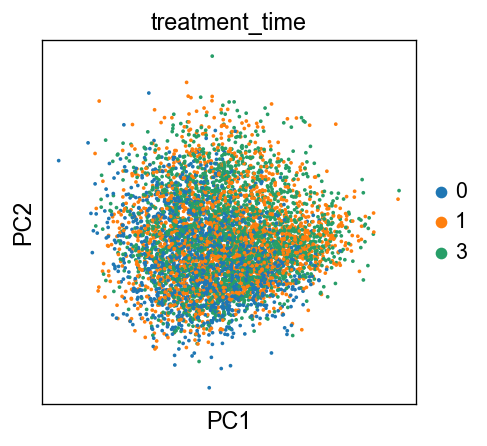
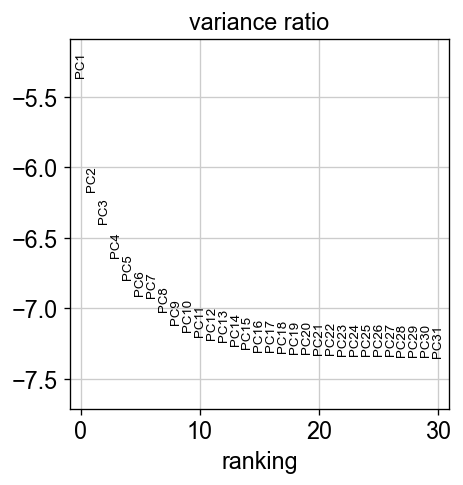
[31]:
sc.pp.neighbors(new_adata, n_pcs=10, use_rep="X_pca")
sc.tl.umap(new_adata)
df = pd.DataFrame(new_adata.obsm['X_umap'], columns=['UMAP1', 'UMAP2'])
df['treatment_time'] = [i + '-hour' for i in new_adata.obs['treatment_time'].tolist()]
df['UMAP1'] = -df['UMAP1']
sns.jointplot(data=df.sort_values(by='treatment_time', ascending=False), x="UMAP1", y="UMAP2", hue="treatment_time", s=4,
palette=[sns.color_palette("tab10")[i] for i in [2, 0, 1]])
computing neighbors
finished: added to `.uns['neighbors']`
`.obsp['distances']`, distances for each pair of neighbors
`.obsp['connectivities']`, weighted adjacency matrix (0:00:00)
computing UMAP
finished: added
'X_umap', UMAP coordinates (adata.obsm) (0:00:07)
[31]:
<seaborn.axisgrid.JointGrid at 0x21b029b7188>

Scatter Plot for All Genes Chosen by SCMER
[32]:
sc.pl.umap(adata, color=adata.var_names[model.get_mask()].tolist(), legend_loc='on data', size=5., color_map='inferno')
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1192: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if is_string_dtype(df[key]) and not is_categorical(df[key])
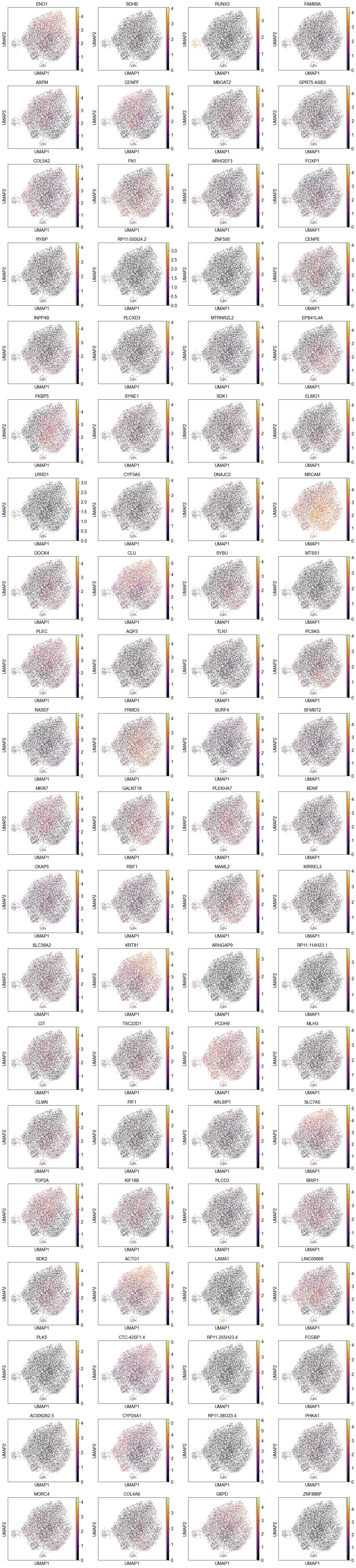
Consensus of RNA and ATAC manifold
[33]:
atac_df = pd.read_pickle("../notebooks/a549-atac-umap.pkl")
[34]:
common_cells = list(set(atac_df.index.tolist()).intersection(adata.obs_names.tolist()))
len(common_cells)
[34]:
1434
[35]:
common_adata = adata[common_cells, :]
common_adata.obsm['X_atac_umap'] = atac_df.loc[common_cells, ['UMAP1', 'UMAP2']].values
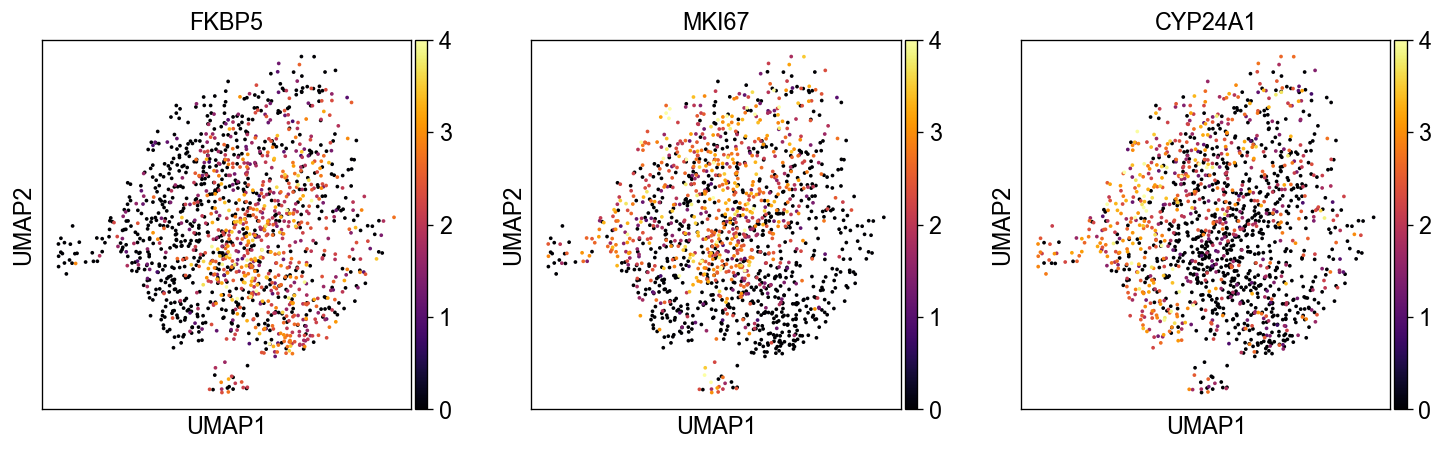
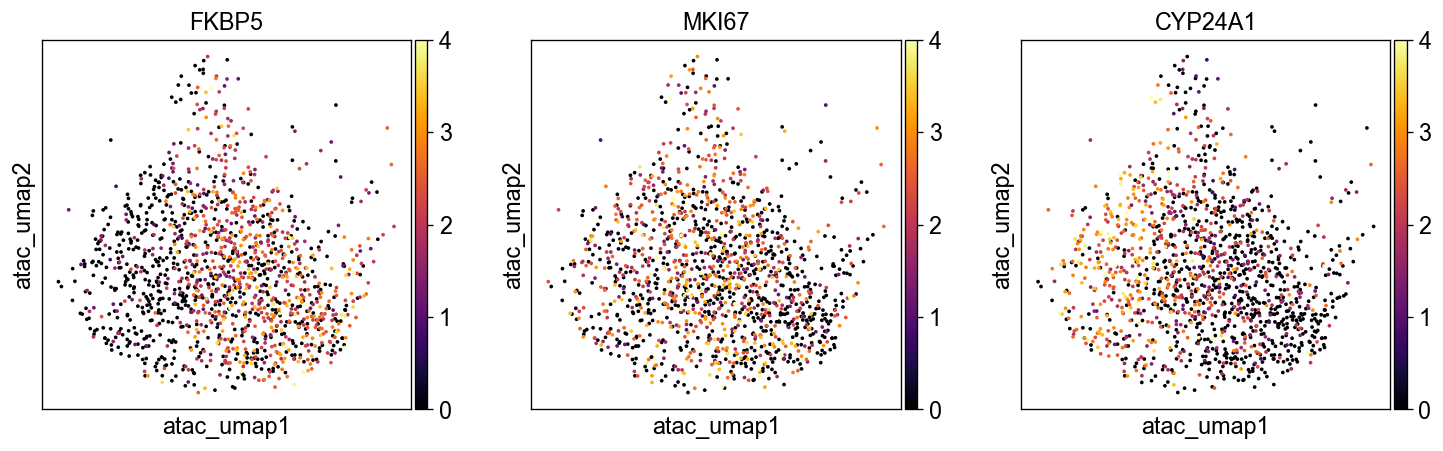
sc.pl.embedding(common_adata, 'umap', color=['FKBP5', 'MKI67', 'CYP24A1'], legend_loc='on data', size=20., ncols=3,
color_map='inferno', vmax=4)
sc.pl.embedding(common_adata, 'atac_umap', color=['FKBP5', 'MKI67', 'CYP24A1'], legend_loc='on data', size=20., ncols=3,
color_map='inferno', vmax=4)
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1094: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if not is_categorical(df_full[k]):
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1192: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if is_string_dtype(df[key]) and not is_categorical(df[key])
[36]:
common_adata = adata[common_cells, :]
common_adata.obsm['X_atac_umap'] = atac_df.loc[common_cells, ['UMAP1', 'UMAP2']].values
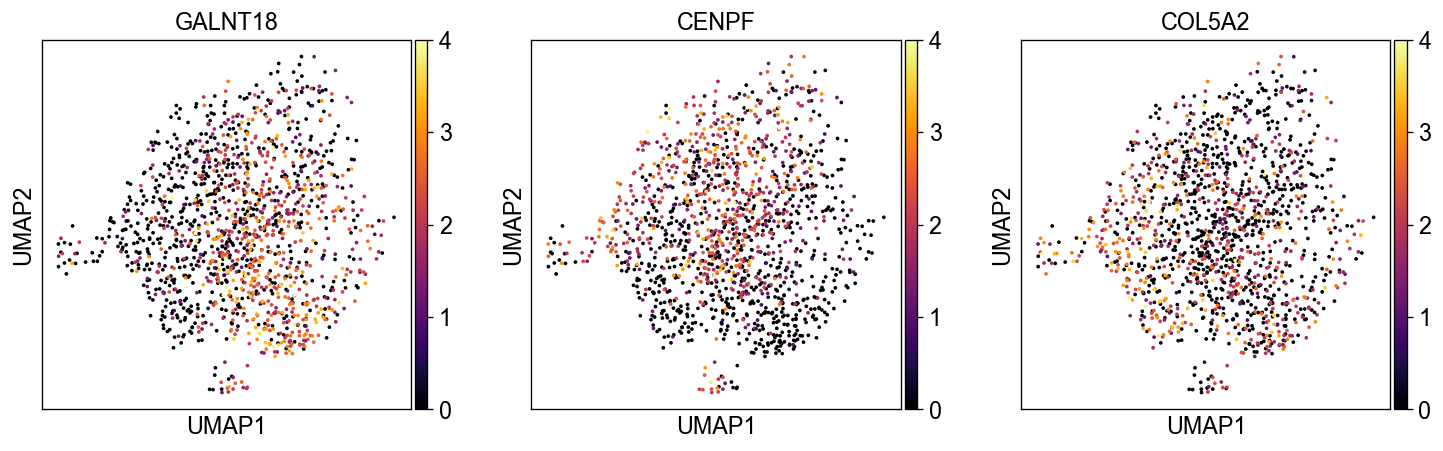
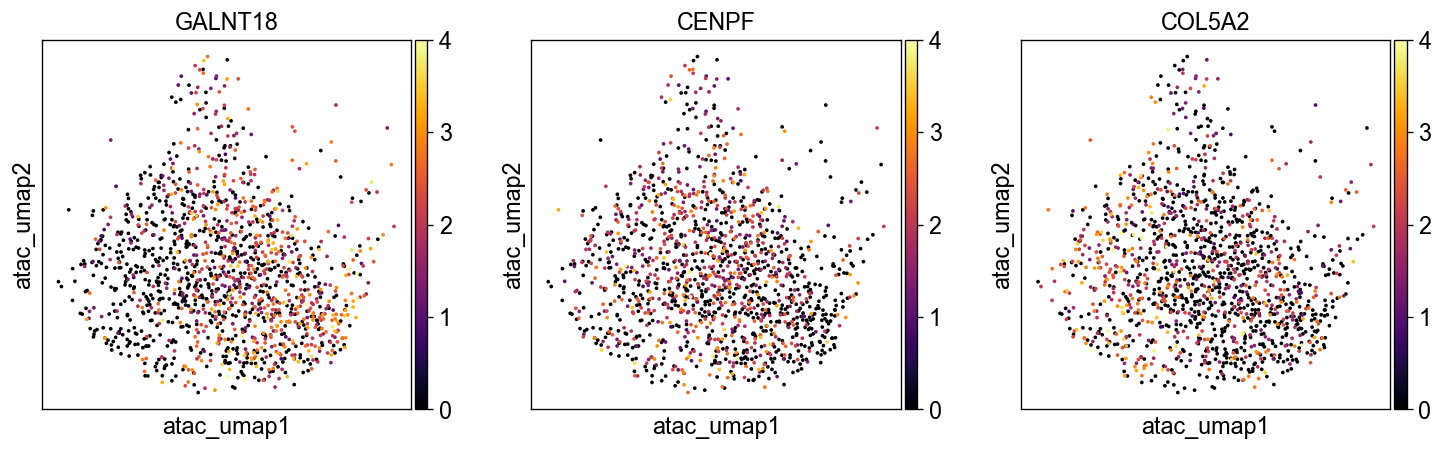
sc.pl.embedding(common_adata, 'umap', color=['GALNT18', 'CENPF', 'COL5A2'], legend_loc='on data', size=20., ncols=3,
color_map='inferno', vmax=4)
sc.pl.embedding(common_adata, 'atac_umap', color=['GALNT18', 'CENPF', 'COL5A2'], legend_loc='on data', size=20., ncols=3,
color_map='inferno', vmax=4)
Correlation with TFs
[37]:
tf = pd.read_csv("../../sci-car/gold_standard_tf.csv", index_col=0).T
tf = tf.loc[common_cells, :]
[38]:
import scipy.stats
[39]:
corr_df = pd.DataFrame([np.corrcoef(np.array(common_adata[:, g].X).squeeze(), tf['MA0113.3_NR3C1'])[0, 1] for g in adata.var_names],
index=adata.var_names, columns=['Correlation'])
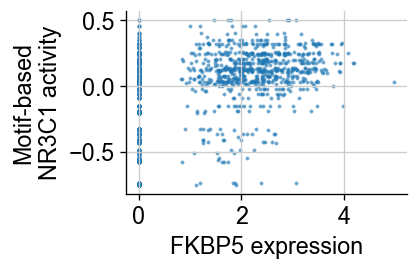
Among all the genes, FKBP5 and CYP24A1 are the most correlated with the activity of NR3C1 TF, a major target of DEX.
[40]:
corr_df['score'] = model.w
corr_df.sort_values(by='Correlation', ascending=False)
[40]:
| Correlation | score | |
|---|---|---|
| gene | ||
| FKBP5 | 0.355375 | 1.584072 |
| GALNT18 | 0.255927 | 0.888890 |
| NRCAM | 0.235787 | 1.165094 |
| SYBU | 0.201692 | 0.845990 |
| PLEKHA7 | 0.201616 | 0.848208 |
| ... | ... | ... |
| KIFC3 | -0.192754 | 0.000000 |
| MAP3K14 | -0.208078 | 0.000000 |
| COL5A2 | -0.218765 | 0.913006 |
| KAZN | -0.223749 | 0.000000 |
| CYP24A1 | -0.365414 | 1.473135 |
6850 rows × 2 columns
[41]:
g = 'FKBP5'
fig = plt.figure(figsize=(3, 2))
plt.scatter(np.array(common_adata.raw.to_adata()[:, g].X.todense()).squeeze(), tf['MA0113.3_NR3C1'], s=2., alpha=.5)
fig.axes[0].spines['right'].set_visible(False)
fig.axes[0].spines['top'].set_visible(False)
plt.ylabel("Motif-based\nNR3C1 activity")
plt.xlabel("FKBP5 expression")
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1094: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if not is_categorical(df_full[k]):
[41]:
Text(0.5, 0, 'FKBP5 expression')
[42]:
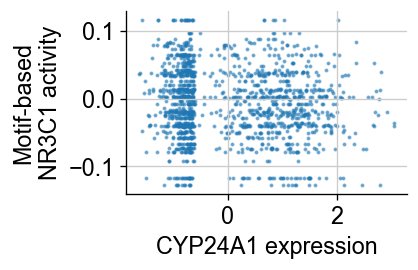
g = 'CYP24A1'
fig = plt.figure(figsize=(3, 2))
plt.scatter(np.array(common_adata.raw.to_adata()[:, g].X.todense()).squeeze(), tf['MA0113.3_NR3C1'], s=2., alpha=.5)
fig.axes[0].spines['right'].set_visible(False)
fig.axes[0].spines['top'].set_visible(False)
plt.ylabel("Motif-based\nNR3C1 activity")
plt.xlabel("CYP24A1 expression")
[42]:
Text(0.5, 0, 'CYP24A1 expression')

The most correlated genes are recalled by SCMER.
[43]:
corr_df[corr_df.score>0.].sort_values(by='score', ascending=False).style
[43]:
| Correlation | score | |
|---|---|---|
| gene | ||
| MKI67 | 0.005315 | 2.196719 |
| CENPF | 0.012876 | 2.181099 |
| TOP2A | 0.005913 | 2.103136 |
| KRT81 | 0.009329 | 1.925768 |
| LINC00669 | -0.026802 | 1.848227 |
| SLC7A5 | -0.033285 | 1.672786 |
| ACTG1 | -0.006641 | 1.635077 |
| FKBP5 | 0.355375 | 1.584072 |
| CYP24A1 | -0.365414 | 1.473135 |
| CENPE | 0.013720 | 1.428560 |
| CLU | -0.076312 | 1.372199 |
| ASPM | -0.008454 | 1.325148 |
| FN1 | 0.004577 | 1.254929 |
| KIF18B | -0.003040 | 1.248316 |
| LRRD1 | -0.147250 | 1.203383 |
| NRCAM | 0.235787 | 1.165094 |
| BRIP1 | 0.004534 | 1.160775 |
| RUNX3 | -0.127338 | 1.158783 |
| PCSK5 | 0.104530 | 1.142940 |
| RP11-255H23.4 | 0.053456 | 1.132451 |
| ENO1 | 0.006445 | 1.121374 |
| LAMA1 | 0.066537 | 1.102744 |
| EPB41L4A | 0.180805 | 1.057562 |
| CTC-425F1.4 | 0.007689 | 1.040152 |
| CIT | 0.023788 | 1.032294 |
| RP11-38O23.4 | 0.027594 | 1.018302 |
| KIRREL3 | 0.002230 | 1.003883 |
| FRMD3 | 0.179904 | 0.998225 |
| ARHGAP9 | -0.092531 | 0.969304 |
| PLEC | -0.008497 | 0.955909 |
| PIF1 | -0.000007 | 0.947894 |
| MAML2 | 0.031461 | 0.931636 |
| INPP4B | -0.000688 | 0.929080 |
| COL5A2 | -0.218765 | 0.913006 |
| G6PD | -0.027089 | 0.894567 |
| GALNT18 | 0.255927 | 0.888890 |
| DOCK4 | 0.161354 | 0.876944 |
| PLEKHA7 | 0.201616 | 0.848208 |
| SYBU | 0.201692 | 0.845990 |
| PCDH9 | -0.025937 | 0.839740 |
| SDK2 | 0.174713 | 0.827627 |
| SDK1 | 0.072312 | 0.796790 |
| CLMN | 0.152751 | 0.761204 |
| AC006262.5 | 0.088222 | 0.715820 |
| SURF4 | 0.022712 | 0.697328 |
| SYNE1 | -0.052182 | 0.686870 |
| AQP3 | 0.075974 | 0.684257 |
| ARL6IP1 | 0.020573 | 0.651571 |
| PLCD3 | -0.017449 | 0.623484 |
| CYP3A5 | 0.040255 | 0.622529 |
| CKAP5 | 0.023530 | 0.606939 |
| COL4A6 | 0.096919 | 0.600528 |
| MORC4 | -0.065141 | 0.559950 |
| PHKA1 | 0.077768 | 0.555157 |
| MTRNR2L2 | -0.024974 | 0.543801 |
| RP11-550I24.2 | -0.081131 | 0.508427 |
| SFMBT2 | -0.032559 | 0.501230 |
| PLCXD3 | -0.022115 | 0.495945 |
| FOXP1 | 0.061419 | 0.449230 |
| RASEF | 0.032687 | 0.446423 |
| DNAJC2 | -0.062482 | 0.437808 |
| RYBP | 0.000711 | 0.435919 |
| MLH3 | 0.033287 | 0.426470 |
| ELMO1 | 0.057909 | 0.424187 |
| RP11-114H23.1 | -0.011697 | 0.423165 |
| SDHB | -0.044482 | 0.421593 |
| BDNF | -0.011787 | 0.417173 |
| FCGBP | -0.046208 | 0.371409 |
| MBOAT2 | 0.134323 | 0.359302 |
| FAM69A | -0.019410 | 0.326981 |
| TLN1 | 0.024994 | 0.301150 |
| SLC38A2 | 0.023776 | 0.280628 |
| ZNF595 | -0.041044 | 0.267564 |
| PLK5 | 0.045267 | 0.256589 |
| RSF1 | -0.015783 | 0.234042 |
| ARHGEF3 | -0.044143 | 0.220962 |
| MTSS1 | 0.031700 | 0.116672 |
| ZNF886P | 0.008182 | 0.070685 |
| GPR75-ASB3 | 0.002737 | 0.029292 |
| TSC22D1 | 0.037347 | 0.029128 |
[44]:
corr_df[corr_df.score>0.].sort_values(by='Correlation', ascending=False).style
[44]:
| Correlation | score | |
|---|---|---|
| gene | ||
| FKBP5 | 0.355375 | 1.584072 |
| GALNT18 | 0.255927 | 0.888890 |
| NRCAM | 0.235787 | 1.165094 |
| SYBU | 0.201692 | 0.845990 |
| PLEKHA7 | 0.201616 | 0.848208 |
| EPB41L4A | 0.180805 | 1.057562 |
| FRMD3 | 0.179904 | 0.998225 |
| SDK2 | 0.174713 | 0.827627 |
| DOCK4 | 0.161354 | 0.876944 |
| CLMN | 0.152751 | 0.761204 |
| MBOAT2 | 0.134323 | 0.359302 |
| PCSK5 | 0.104530 | 1.142940 |
| COL4A6 | 0.096919 | 0.600528 |
| AC006262.5 | 0.088222 | 0.715820 |
| PHKA1 | 0.077768 | 0.555157 |
| AQP3 | 0.075974 | 0.684257 |
| SDK1 | 0.072312 | 0.796790 |
| LAMA1 | 0.066537 | 1.102744 |
| FOXP1 | 0.061419 | 0.449230 |
| ELMO1 | 0.057909 | 0.424187 |
| RP11-255H23.4 | 0.053456 | 1.132451 |
| PLK5 | 0.045267 | 0.256589 |
| CYP3A5 | 0.040255 | 0.622529 |
| TSC22D1 | 0.037347 | 0.029128 |
| MLH3 | 0.033287 | 0.426470 |
| RASEF | 0.032687 | 0.446423 |
| MTSS1 | 0.031700 | 0.116672 |
| MAML2 | 0.031461 | 0.931636 |
| RP11-38O23.4 | 0.027594 | 1.018302 |
| TLN1 | 0.024994 | 0.301150 |
| CIT | 0.023788 | 1.032294 |
| SLC38A2 | 0.023776 | 0.280628 |
| CKAP5 | 0.023530 | 0.606939 |
| SURF4 | 0.022712 | 0.697328 |
| ARL6IP1 | 0.020573 | 0.651571 |
| CENPE | 0.013720 | 1.428560 |
| CENPF | 0.012876 | 2.181099 |
| KRT81 | 0.009329 | 1.925768 |
| ZNF886P | 0.008182 | 0.070685 |
| CTC-425F1.4 | 0.007689 | 1.040152 |
| ENO1 | 0.006445 | 1.121374 |
| TOP2A | 0.005913 | 2.103136 |
| MKI67 | 0.005315 | 2.196719 |
| FN1 | 0.004577 | 1.254929 |
| BRIP1 | 0.004534 | 1.160775 |
| GPR75-ASB3 | 0.002737 | 0.029292 |
| KIRREL3 | 0.002230 | 1.003883 |
| RYBP | 0.000711 | 0.435919 |
| PIF1 | -0.000007 | 0.947894 |
| INPP4B | -0.000688 | 0.929080 |
| KIF18B | -0.003040 | 1.248316 |
| ACTG1 | -0.006641 | 1.635077 |
| ASPM | -0.008454 | 1.325148 |
| PLEC | -0.008497 | 0.955909 |
| RP11-114H23.1 | -0.011697 | 0.423165 |
| BDNF | -0.011787 | 0.417173 |
| RSF1 | -0.015783 | 0.234042 |
| PLCD3 | -0.017449 | 0.623484 |
| FAM69A | -0.019410 | 0.326981 |
| PLCXD3 | -0.022115 | 0.495945 |
| MTRNR2L2 | -0.024974 | 0.543801 |
| PCDH9 | -0.025937 | 0.839740 |
| LINC00669 | -0.026802 | 1.848227 |
| G6PD | -0.027089 | 0.894567 |
| SFMBT2 | -0.032559 | 0.501230 |
| SLC7A5 | -0.033285 | 1.672786 |
| ZNF595 | -0.041044 | 0.267564 |
| ARHGEF3 | -0.044143 | 0.220962 |
| SDHB | -0.044482 | 0.421593 |
| FCGBP | -0.046208 | 0.371409 |
| SYNE1 | -0.052182 | 0.686870 |
| DNAJC2 | -0.062482 | 0.437808 |
| MORC4 | -0.065141 | 0.559950 |
| CLU | -0.076312 | 1.372199 |
| RP11-550I24.2 | -0.081131 | 0.508427 |
| ARHGAP9 | -0.092531 | 0.969304 |
| RUNX3 | -0.127338 | 1.158783 |
| LRRD1 | -0.147250 | 1.203383 |
| COL5A2 | -0.218765 | 0.913006 |
| CYP24A1 | -0.365414 | 1.473135 |
MKI67, an important cell cycle marker, is uncorrelated with NR3C1.
[45]:
g = 'MKI67'
fig = plt.figure(figsize=(3, 2))
plt.scatter(np.array(common_adata[:, g].X).squeeze(), tf['MA0106.3_TP53'], s=2., alpha=.5)
fig.axes[0].spines['right'].set_visible(False)
fig.axes[0].spines['top'].set_visible(False)
plt.ylabel("Motif-based\nNR3C1 activity")
plt.xlabel("CYP24A1 expression")
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1094: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if not is_categorical(df_full[k]):
[45]:
Text(0.5, 0, 'CYP24A1 expression')
[46]:
corr_df2 = pd.DataFrame([np.corrcoef(np.array(common_adata[:, 'MKI67'].X).squeeze(), tf[t])[0, 1] for t in tf.columns],
index=tf.columns, columns=['Correlation'])
corr_df2.sort_values(by="Correlation")
[46]:
| Correlation | |
|---|---|
| MA0106.3_TP53 | -0.098055 |
| MA0665.1_MSC | -0.091495 |
| MA0698.1_ZBTB18 | -0.088254 |
| MA0148.3_FOXA1 | -0.087623 |
| MA0140.2_GATA1::TAL1 | -0.085941 |
| ... | ... |
| MA0773.1_MEF2D | 0.096405 |
| MA0749.1_ZBED1 | 0.097567 |
| MA0788.1_POU3F3 | 0.099256 |
| MA0862.1_GMEB2 | 0.107898 |
| MA0753.1_ZNF740 | 0.117872 |
386 rows × 1 columns