Ileum Lamina Propria Immunocyte Subtypes (Martin et al.)
Dataset download: Immune Cell Atlas
[1]:
import scanpy as sc
import os
import pandas as pd
import numpy as np
import pickle as pkl
import matplotlib as mpl
import matplotlib.pyplot as plt
import scipy.stats
[2]:
sc.settings.verbosity = 3 # verbosity: errors (0), warnings (1), info (2), hints (3)
sc.settings.n_jobs = 6
Load data
[3]:
adata = sc.read_10x_mtx("../../Ileum/", cache=True)
adata
... reading from cache file cache\..-..-Ileum-matrix.h5ad
[3]:
AnnData object with n_obs × n_vars = 39671 × 33660
var: 'gene_ids'
[4]:
obs = pd.read_table("../../Ileum/Effi_tsne_final.txt", header=[0, 1])
obs.columns = [i[0] for i in obs.columns]
obs.index = obs.NAME
adata = adata[obs.NAME, :]
adata.obs = obs
adata.obs
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1094: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if not is_categorical(df_full[k]):
[4]:
| NAME | X | Y | Primary Lineages | kmeans | |
|---|---|---|---|---|---|
| NAME | |||||
| 122_AAACCTGGTTCCCGAG | 122_AAACCTGGTTCCCGAG | -17.523981 | 25.760201 | IgA plasma cells | 5 |
| 122_AAAGATGTCAGCTCTC | 122_AAAGATGTCAGCTCTC | -4.299636 | -1.107461 | Tregs | 2 |
| 122_AAAGCAACAAGAAGAG | 122_AAAGCAACAAGAAGAG | 18.438143 | -1.855235 | Mast cells | 17 |
| 122_AAATGCCTCACCCGAG | 122_AAATGCCTCACCCGAG | 8.266746 | 4.860057 | TRM | 3 |
| 122_AACCATGCATCACCCT | 122_AACCATGCATCACCCT | 1.359606 | 36.710104 | IgA plasma cells | 10 |
| ... | ... | ... | ... | ... | ... |
| 209_TTTGTCAGTCTCTTAT | 209_TTTGTCAGTCTCTTAT | 11.309222 | 21.416638 | TRM | 1 |
| 209_TTTGTCAGTGTGGTTT | 209_TTTGTCAGTGTGGTTT | 23.971407 | 1.429820 | Group3 ILC | 13 |
| 209_TTTGTCATCAGTTAGC | 209_TTTGTCATCAGTTAGC | 8.514284 | 0.595697 | TRM | 3 |
| 209_TTTGTCATCGTCCAGG | 209_TTTGTCATCGTCCAGG | 14.008064 | 20.373886 | TRM | 1 |
| 209_TTTGTCATCGTTTGCC | 209_TTTGTCATCGTTTGCC | 21.302798 | 7.139810 | TRM | 1 |
39563 rows × 5 columns
[5]:
adata.obs['batch'] = adata.obs.NAME.str[0:3]
print(*adata.obs['batch'].unique())
122 128 138 158 181 187 190 193 196 209
SCANPY Analysis
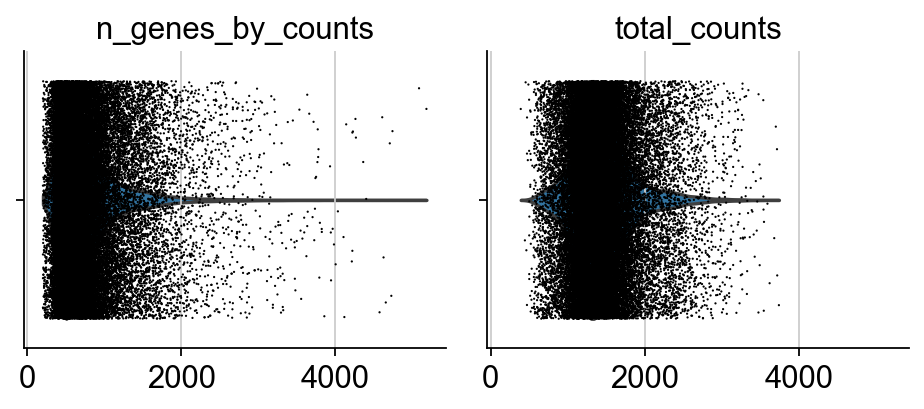
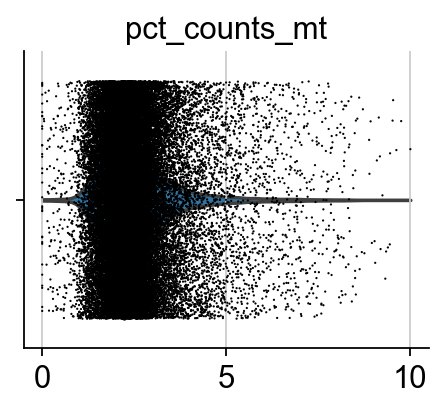
The dataset is already filtered, so no need to do additional QC. Nevertheless, we show the figures.
[6]:
sc.settings.set_figure_params(dpi=80, facecolor='white')
sc.pl.highest_expr_genes(adata, n_top=20)
normalizing counts per cell
finished (0:00:00)

[7]:
adata.var['mt'] = adata.var.index.str.startswith('MT-') # annotate the group of mitochondrial genes as 'mt'
sc.pp.calculate_qc_metrics(adata, qc_vars=['mt'], percent_top=None, log1p=False, inplace=True)
sc.pl.violin(adata, ['n_genes_by_counts', 'total_counts'], jitter=0.4, multi_panel=True)
sc.pl.violin(adata, ['pct_counts_mt'], jitter=0.4, multi_panel=True)
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1192: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if is_string_dtype(df[key]) and not is_categorical(df[key])
... storing 'Primary Lineages' as categorical
... storing 'batch' as categorical
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\seaborn\_core.py:1303: UserWarning: Vertical orientation ignored with only `x` specified.
warnings.warn(single_var_warning.format("Vertical", "x"))
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\seaborn\_core.py:1303: UserWarning: Vertical orientation ignored with only `x` specified.
warnings.warn(single_var_warning.format("Vertical", "x"))
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\seaborn\_core.py:1303: UserWarning: Vertical orientation ignored with only `x` specified.
warnings.warn(single_var_warning.format("Vertical", "x"))
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\seaborn\_core.py:1303: UserWarning: Vertical orientation ignored with only `x` specified.
warnings.warn(single_var_warning.format("Vertical", "x"))
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1192: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if is_string_dtype(df[key]) and not is_categorical(df[key])
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\seaborn\_core.py:1303: UserWarning: Vertical orientation ignored with only `x` specified.
warnings.warn(single_var_warning.format("Vertical", "x"))
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\seaborn\_core.py:1303: UserWarning: Vertical orientation ignored with only `x` specified.
warnings.warn(single_var_warning.format("Vertical", "x"))
[8]:
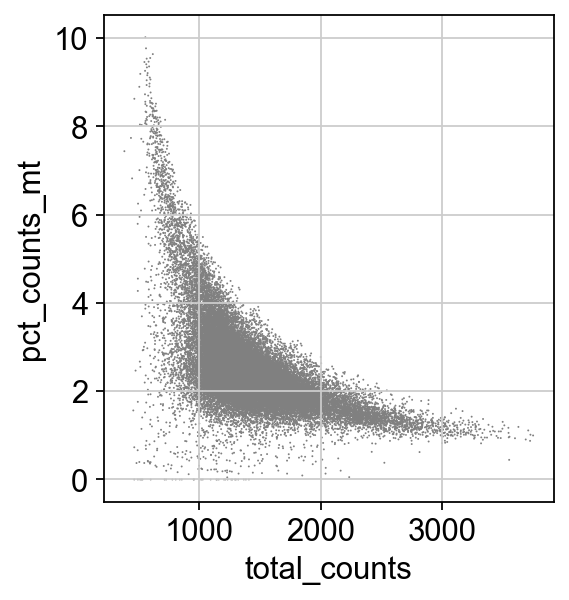
sc.pl.scatter(adata, x='total_counts', y='pct_counts_mt')
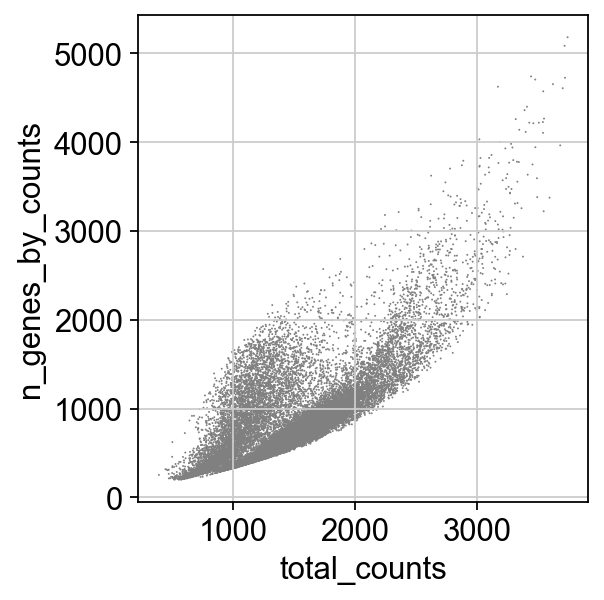
sc.pl.scatter(adata, x='total_counts', y='n_genes_by_counts')
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1192: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if is_string_dtype(df[key]) and not is_categorical(df[key])
[9]:
adata.raw = adata
[10]:
sc.pp.filter_genes(adata, min_cells=3)
filtered out 11319 genes that are detected in less than 3 cells
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1094: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if not is_categorical(df_full[k]):
[11]:
sc.pp.normalize_total(adata, target_sum=1e4)
sc.pp.log1p(adata)
normalizing counts per cell
finished (0:00:00)
[12]:
sc.pp.highly_variable_genes(adata)
sc.pl.highly_variable_genes(adata)
extracting highly variable genes
finished (0:00:00)
--> added
'highly_variable', boolean vector (adata.var)
'means', float vector (adata.var)
'dispersions', float vector (adata.var)
'dispersions_norm', float vector (adata.var)

[13]:
adata = adata[:, adata.var.highly_variable]
adata
[13]:
View of AnnData object with n_obs × n_vars = 39563 × 3573
obs: 'NAME', 'X', 'Y', 'Primary Lineages', 'kmeans', 'batch', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt'
var: 'gene_ids', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'n_cells', 'highly_variable', 'means', 'dispersions', 'dispersions_norm'
uns: 'log1p', 'hvg'
[14]:
sc.pp.scale(adata, max_value=10)
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\scanpy\preprocessing\_simple.py:806: UserWarning: Revieved a view of an AnnData. Making a copy.
view_to_actual(adata)
... as `zero_center=True`, sparse input is densified and may lead to large memory consumption
[ ]:
[15]:
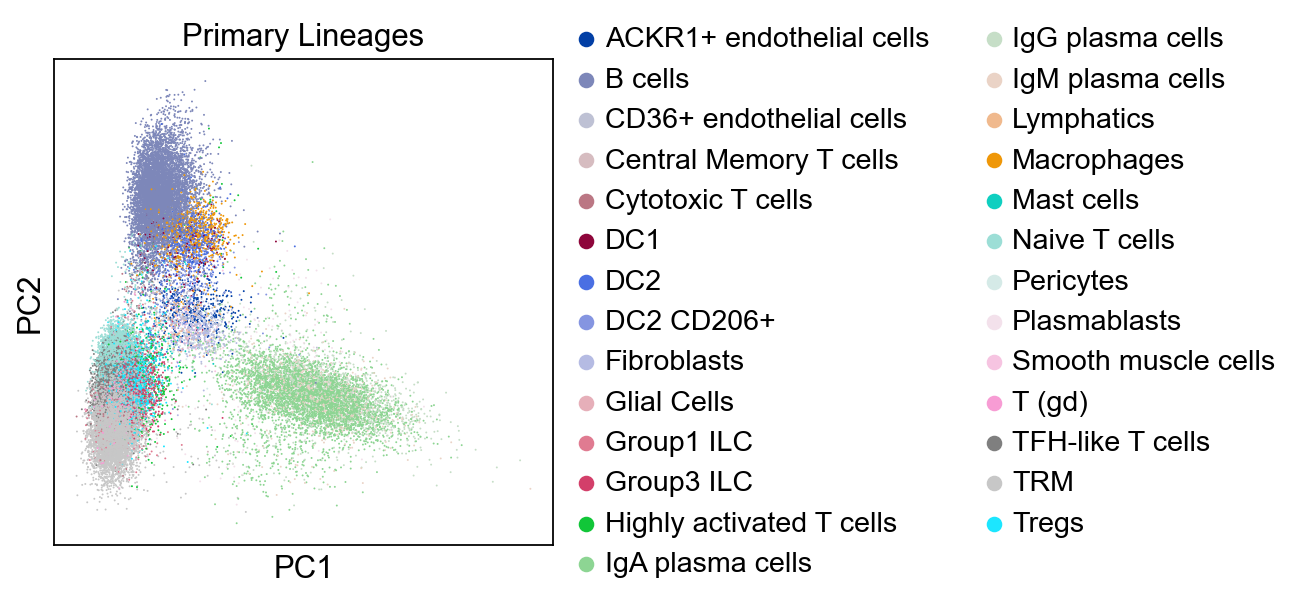
sc.tl.pca(adata, svd_solver='arpack')
sc.pl.pca(adata, color="Primary Lineages")
computing PCA
on highly variable genes
with n_comps=50
finished (0:00:11)
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1192: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if is_string_dtype(df[key]) and not is_categorical(df[key])
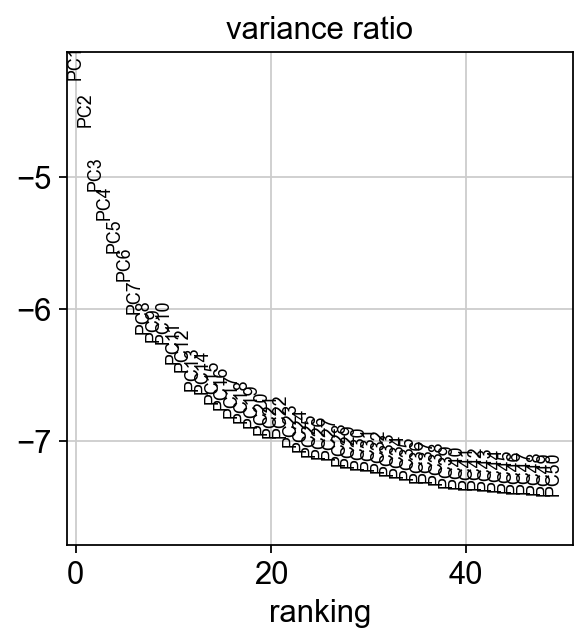
[16]:
sc.pl.pca_variance_ratio(adata, log=True, n_pcs=50)
[17]:
# sc.tl.rank_genes_groups(adata, 'Primary Lineages', method='wilcoxon')
# sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False, ncols=3)
[18]:
sc.pp.neighbors(adata)
sc.tl.umap(adata)
computing neighbors
using 'X_pca' with n_pcs = 50
finished: added to `.uns['neighbors']`
`.obsp['distances']`, distances for each pair of neighbors
`.obsp['connectivities']`, weighted adjacency matrix (0:00:17)
computing UMAP
finished: added
'X_umap', UMAP coordinates (adata.obsm) (0:00:25)
[19]:
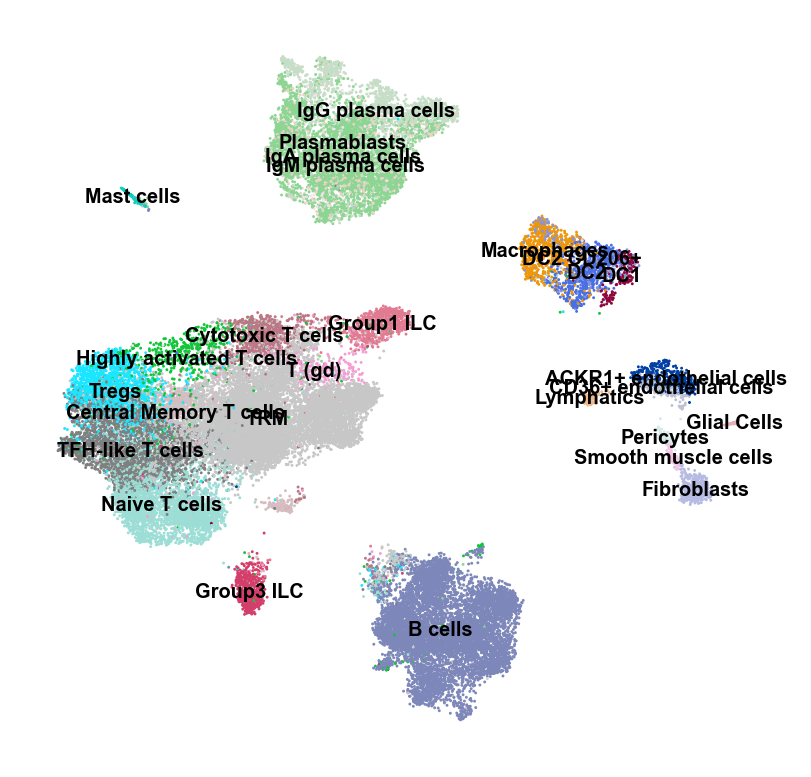
sc.settings.set_figure_params(dpi=120, facecolor='white')
sc.pl.umap(adata, color="Primary Lineages", legend_loc="on data", legend_fontsize=6, frameon=False, title="")
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1192: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if is_string_dtype(df[key]) and not is_categorical(df[key])
SCMER Marker Selection
[20]:
#%autoreload
import sys
sys.path.insert(0,'..')
import scmer
model = scmer.UmapL1(w=1., lasso=2.45e-4, ridge=2.45e-4, n_pcs=50, perplexity=100., use_beta_in_Q=True, n_threads=6,
max_outer_iter=2)
model.fit(adata.X, batches=adata.obs['batch'].values)
Batch 122 with 437 instances.
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 1.284627
Done. Elapsed time: 2.49 seconds. Total: 2.49 seconds.
Batch 128 with 2312 instances.
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 1.006885
Done. Elapsed time: 6.60 seconds. Total: 9.09 seconds.
Batch 138 with 3830 instances.
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 0.873896
Done. Elapsed time: 13.12 seconds. Total: 22.21 seconds.
Batch 158 with 3132 instances.
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 0.890955
Done. Elapsed time: 9.92 seconds. Total: 32.13 seconds.
Batch 181 with 8782 instances.
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 0.716225
Done. Elapsed time: 60.60 seconds. Total: 92.72 seconds.
Batch 187 with 892 instances.
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 1.215405
Done. Elapsed time: 2.97 seconds. Total: 95.69 seconds.
Batch 190 with 4759 instances.
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 0.791321
Done. Elapsed time: 19.27 seconds. Total: 114.96 seconds.
Batch 193 with 8833 instances.
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 0.742579
Done. Elapsed time: 62.02 seconds. Total: 176.98 seconds.
Batch 196 with 2477 instances.
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 0.992064
Done. Elapsed time: 7.13 seconds. Total: 184.11 seconds.
Batch 209 with 4109 instances.
Calculating distance matrix and scaling factors...
Computing pairwise distances...
Using 6 threads...
Mean value of sigma: 0.863905
Done. Elapsed time: 15.84 seconds. Total: 199.95 seconds.
Creating model without batches...
Optimizing using OWLQN (because lasso is nonzero)...
0 loss: 4.905979633331299 Nonzero: 259 Elapsed time: 714.81 seconds. Total: 914.76 seconds.
1 loss: 2.8209409713745117 Nonzero: 250 Elapsed time: 491.56 seconds. Total: 1406.32 seconds.
final loss: 2.788534164428711 Nonzero: 250 Elapsed time: 11.33 seconds. Total: 1417.65 seconds.
[20]:
<scmer._umap_l1.UmapL1 at 0x1639ec51fc8>
[21]:
selected_markers = adata.var_names[model.w > 0]
all_markers = adata.var_names
[22]:
print(*selected_markers, sep=', ')
TNFRSF18, TNFRSF4, C1QA, C1QC, C1QB, ID3, LAPTM5, MARCKSL1, JUN, RP5-887A10.1, ODF2L, CHI3L2, SLC16A1-AS1, CD160, TXNIP, ACKR1, FCER1G, HSPA6, XCL2, XCL1, SELL, RGS1, ELF3, PIGR, SLC30A1, MTRNR2L11, AC092580.4, LINC00299, ID2, THADA, REG1B, REG1A, TRABD2A, GNLY, CD8A, CD8B, LINC00152, FABP1, IGKV1-12, IGKV3-15, DUSP2, MIR4435-2HG, IL1B, CXCR4, COL3A1, AC005540.3, STAT4, PGAP1, CD28, CTLA4, ICOS, CCL20, ITM2C, PASK, ANKRD28, CMC1, TRAT1, CD200, TIGIT, GPR171, RP11-3P17.5, KLHL6-AS1, CC2D2A, SMIM14, KIT, HOPX, IGFBP7, JCHAIN, AREG, CXCL13, PLAC8, SPARCL1, SPP1, BANK1, LEF1, FABP2, SPRY1, RP11-18H21.1, 1-Mar.1, IL7R, PTGER4, SEPP1, ITGA1, GZMK, GZMA, PDE4D, MZB1, CD74, FABP6, DUSP1, ADTRP, CD83, FAM65B, AIF1, HLA-DRA, HLA-DRB5, HLA-DRB1, HLA-DQA1, HLA-DQB1, HLA-DQB2, HLA-DMB, HLA-DMA, HLA-DPA1, HLA-DPB1, IL17A, AIM1, SESN1, FYN, TNFAIP3, CITED2, SYTL3, CCR6, ICA1, AGR2, IL6, CLDN4, COL1A2, RP11-138A9.1, CALD1, TRBC1, TRBC2, GIMAP7, PRDX4, SAT1, ZFX, SSR4, DEFA5, ADAMDEC1, DUSP4, RP11-51J9.5, RNF19A, SPINK4, AC129778.2, ANXA1, LCN2, QSOX2, MTRNR2L8, WT1-AS, MS4A1, CYB561A3, FKBP2, NEAT1, CTSW, POU2AF1, BCO2, IL2RA, CREM, RTKN2, DMBT1, RP11-298J20.3, PRAP1, KLRB1, CD69, KLRF1, KLRD1, KLRC1, RP11-291B21.2, FKBP11, IL22, LYZ, PHLDA1, NTS, LUM, DCN, RP11-693J15.5, HSP90B1, UBC, ZNF10, RGCC, KIAA0226L, OLFM4, LINC00402, TBC1D4, RNASE6, TRDC, GZMH, GZMB, AC005480.1, FOS, BATF, TC2N, IFI27, TCL1A, IGHA2, IGHG4, IGHG2, IGHGP, IGHG1, IGHG3, IGHD, IGHV1-2, IGHV3-74, LINC00926, RORA, TPSB2, TPSAB1, IL32, TNFRSF17, MT2A, MT1E, MT1X, HERPUD1, MAF, IRF8, CYBA, CCL11, CCL5, CCL4, CCL4L2, RP5-1028K7.2, CCR7, CD79B, SNHG25, CD7, SEC11C, CST3, CST7, TSHZ2, FCER2, JUNB, ZSWIM4, DNAJB1, ZNF90, TYROBP, ZFP36, CD79A, NKG7, ZFY, TTTY15, USP9Y, UTY, KDM5D, AC008079.10, IGLV1-51, IGLV1-41, IGLV2-23, IGLV3-19, IGLV2-11, IGLC2, IGLC5, IGLC6, IGLC7, IGLL1, VPREB3, DERL3, XBP1, LGALS1, FAM118A, CH507-42P11.8, TFF3
Validation
PCA and UMAP
[23]:
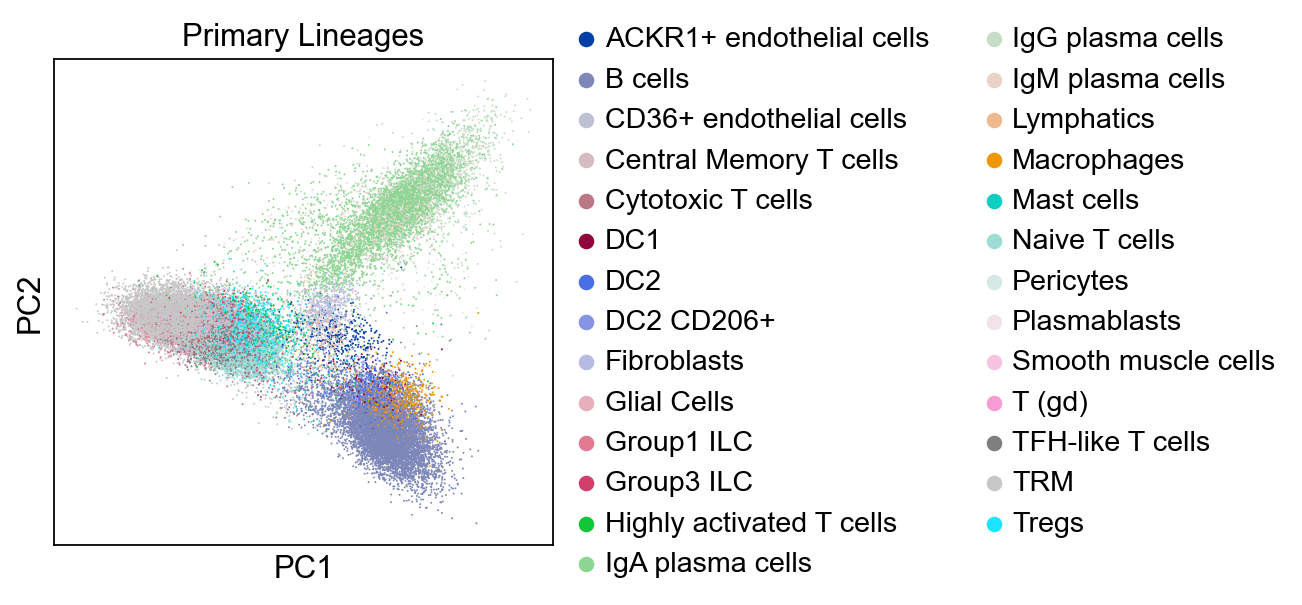
new_adata = model.transform(adata)
sc.tl.pca(new_adata, svd_solver='arpack')
sc.settings.set_figure_params(dpi=80, facecolor='white')
sc.pl.pca(new_adata, color="Primary Lineages")
sc.pp.neighbors(new_adata, n_pcs=20)
sc.tl.umap(new_adata)
sc.settings.set_figure_params(dpi=120, facecolor='white')
sc.pl.umap(new_adata, color="Primary Lineages", legend_loc="on data", legend_fontsize=6, frameon=False)
computing PCA
on highly variable genes
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1094: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if not is_categorical(df_full[k]):
with n_comps=50
finished (0:00:01)
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1192: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if is_string_dtype(df[key]) and not is_categorical(df[key])
computing neighbors
using 'X_pca' with n_pcs = 20
finished: added to `.uns['neighbors']`
`.obsp['distances']`, distances for each pair of neighbors
`.obsp['connectivities']`, weighted adjacency matrix (0:00:06)
computing UMAP
finished: added
'X_umap', UMAP coordinates (adata.obsm) (0:00:23)
C:\Users\SLiang3\Miniconda3\envs\scanpy37\lib\site-packages\anndata\_core\anndata.py:1192: FutureWarning: is_categorical is deprecated and will be removed in a future version. Use is_categorical_dtype instead
if is_string_dtype(df[key]) and not is_categorical(df[key])
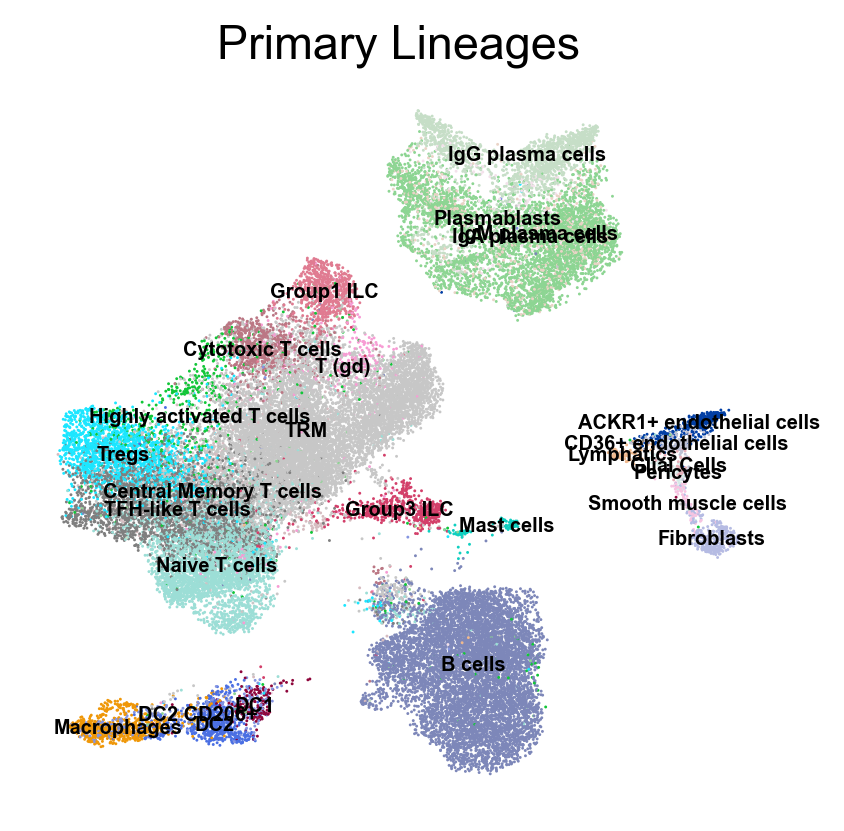
Gene sets curated from the original publication
[24]:
markers = {}
with open("../../Ileum/genes.txt", 'r') as file:
for i in file.readlines():
c, g = i.split(": ")
markers[c] = g.strip().split(', ')
[25]:
cnt = 0
recall_dict = []
for i in markers:
temp = set(markers[i]).intersection(set(selected_markers))
recall_dict.append([i, len(markers[i]), len(set(markers[i]).intersection(set(all_markers))), len(temp), ", ".join(temp)])
recall_dict = pd.DataFrame(recall_dict)
recall_dict.columns = ['Gene set', 'All', 'Highly variable', 'ManMark', 'Recalled Genes']
recall_dict.index = recall_dict['Gene set']
recall_dict.drop('Gene set', axis=1).style
[25]:
| All | Highly variable | ManMark | Recalled Genes | |
|---|---|---|---|---|
| Gene set | ||||
| T cells | 5 | 1 | 1 | CD7 |
| cell cycle | 49 | 0 | 0 | |
| immunoregulation | 15 | 11 | 7 | TIGIT, IL2RA, ICA1, TBC1D4, TNFRSF4, CTLA4, BATF |
| naive/central memory | 7 | 6 | 3 | SELL, LEF1, CCR7 |
| CD8/cytotoxic | 35 | 27 | 18 | CD8A, XCL1, CD160, FCER1G, KLRD1, CCL5, CMC1, NKG7, XCL2, KLRC1, GNLY, GZMA, KLRF1, CD8B, CCL4, CTSW, GZMB, CCL4L2 |
| resident memory | 31 | 21 | 17 | TNFAIP3, CD69, PDE4D, SPRY1, AC092580.4, ANXA1, KLRB1, ANKRD28, IL7R, SYTL3, ID2, STAT4, RORA, JUN, FOS, GPR171, PTGER4 |
| T vs ILC | 11 | 5 | 4 | TPSAB1, LYZ, TNFRSF17, MS4A1 |
| ILC | 10 | 7 | 4 | FCER1G, TYROBP, CD7, KLRB1 |
| ILC1 including NK | 4 | 2 | 1 | IL7R |
| ILC3 | 9 | 3 | 2 | IL22, RORA |
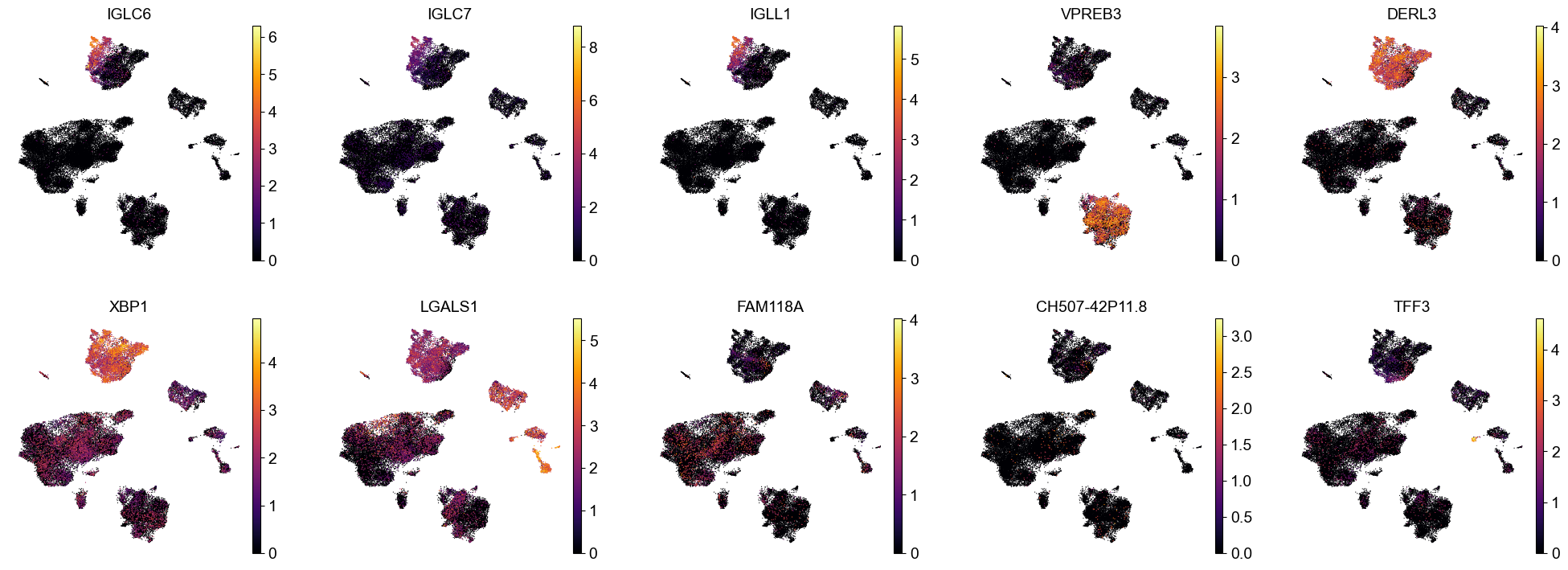
| PC | 12 | 8 | 8 | LGALS1, FKBP2, DERL3, SEC11C, SSR4, FKBP11, HSP90B1, XBP1 |
| IgG | 1 | 1 | 1 | CXCR4 |
| IgM and IgA | 1 | 1 | 1 | TNFRSF17 |
| B | 21 | 19 | 15 | CD79A, HLA-DPB1, HLA-DQB1, BANK1, HLA-DRB5, HLA-DPA1, CD74, HLA-DMB, HLA-DRB1, MS4A1, HLA-DQA1, HLA-DMA, HLA-DRA, CD79B, HLA-DQB2 |
| B naive | 3 | 3 | 2 | FCER2, IGHD |
| B switched memory | 2 | 1 | 1 | IGHG1 |
| Mast | 3 | 3 | 2 | KIT, TPSAB1 |
| Epithelial | 42 | 20 | 15 | FABP1, IFI27, REG1A, TFF3, PIGR, REG1B, DEFA5, FABP2, FABP6, CLDN4, ELF3, AGR2, LCN2, OLFM4, SPINK4 |
| DC | 1 | 0 | 0 | |
| DC2 | 5 | 0 | 0 | |
| DC1 | 6 | 1 | 0 | |
| Macrophages | 24 | 12 | 5 | C1QA, C1QB, C1QC, CCL4, CCL4L2 |
| gut-resident macrophages | 6 | 4 | 3 | C1QA, C1QC, C1QB |
| inflammatory macrophages | 10 | 7 | 2 | CCL4L2, CCL4 |
| lymphatic | 4 | 1 | 0 | |
| subset of blood endothelial | 3 | 1 | 1 | ACKR1 |
| Pericytes | 3 | 1 | 0 | |
| smooth muscle | 4 | 1 | 0 | |
| Fibroblasts | 5 | 2 | 1 | LUM |
| fibroblasts subtype expressed a hallmark activation | 12 | 4 | 1 | IL6 |
| Glial | 1 | 1 | 0 |
[26]:
cnt = 0
for i in markers:
temp = set(markers[i]).intersection(set(selected_markers))
print(i, len(temp), '/', len(set(markers[i]).intersection(set(all_markers))), "(", len(markers[i]), "):", end="")
print(*temp)
if len(temp) > 0:
cnt += 1
print()
print('Recalled', cnt, 'in', len(markers), 'gene sets.')
T cells 1 / 1 ( 5 ):CD7
cell cycle 0 / 0 ( 49 ):
immunoregulation 7 / 11 ( 15 ):TIGIT IL2RA ICA1 TBC1D4 TNFRSF4 CTLA4 BATF
naive/central memory 3 / 6 ( 7 ):SELL LEF1 CCR7
CD8/cytotoxic 18 / 27 ( 35 ):CD8A XCL1 CD160 FCER1G KLRD1 CCL5 CMC1 NKG7 XCL2 KLRC1 GNLY GZMA KLRF1 CD8B CCL4 CTSW GZMB CCL4L2
resident memory 17 / 21 ( 31 ):TNFAIP3 CD69 PDE4D SPRY1 AC092580.4 ANXA1 KLRB1 ANKRD28 IL7R SYTL3 ID2 STAT4 RORA JUN FOS GPR171 PTGER4
T vs ILC 4 / 5 ( 11 ):TPSAB1 LYZ TNFRSF17 MS4A1
ILC 4 / 7 ( 10 ):FCER1G TYROBP CD7 KLRB1
ILC1 including NK 1 / 2 ( 4 ):IL7R
ILC3 2 / 3 ( 9 ):IL22 RORA
PC 8 / 8 ( 12 ):LGALS1 FKBP2 DERL3 SEC11C SSR4 FKBP11 HSP90B1 XBP1
IgG 1 / 1 ( 1 ):CXCR4
IgM and IgA 1 / 1 ( 1 ):TNFRSF17
B 15 / 19 ( 21 ):CD79A HLA-DPB1 HLA-DQB1 BANK1 HLA-DRB5 HLA-DPA1 CD74 HLA-DMB HLA-DRB1 MS4A1 HLA-DQA1 HLA-DMA HLA-DRA CD79B HLA-DQB2
B naive 2 / 3 ( 3 ):FCER2 IGHD
B switched memory 1 / 1 ( 2 ):IGHG1
Mast 2 / 3 ( 3 ):KIT TPSAB1
Epithelial 15 / 20 ( 42 ):FABP1 IFI27 REG1A TFF3 PIGR REG1B DEFA5 FABP2 FABP6 CLDN4 ELF3 AGR2 LCN2 OLFM4 SPINK4
DC 0 / 0 ( 1 ):
DC2 0 / 0 ( 5 ):
DC1 0 / 1 ( 6 ):
Macrophages 5 / 12 ( 24 ):C1QA C1QB C1QC CCL4 CCL4L2
gut-resident macrophages 3 / 4 ( 6 ):C1QA C1QC C1QB
inflammatory macrophages 2 / 7 ( 10 ):CCL4L2 CCL4
lymphatic 0 / 1 ( 4 ):
subset of blood endothelial 1 / 1 ( 3 ):ACKR1
Pericytes 0 / 1 ( 3 ):
smooth muscle 0 / 1 ( 4 ):
Fibroblasts 1 / 2 ( 5 ):LUM
fibroblasts subtype expressed a hallmark activation 1 / 4 ( 12 ):IL6
Glial 0 / 1 ( 1 ):
Recalled 23 in 31 gene sets.
Genes not in any of these gene sets:
[27]:
temp = set(selected_markers.copy())
for i in markers:
temp -= set(markers[i])
print(*temp, sep=', ')
ITGA1, IGLV2-23, CD200, ZFP36, IRF8, SLC30A1, CHI3L2, AREG, ADTRP, SESN1, SPP1, CXCL13, IGHV3-74, MAF, CD83, RP11-51J9.5, IGLV3-19, IGLC7, COL3A1, LINC00299, LINC00152, RP11-138A9.1, SAT1, RP11-298J20.3, QSOX2, IGKV1-12, SMIM14, RTKN2, MT2A, PASK, IGLL1, CYB561A3, WT1-AS, KIAA0226L, CITED2, CCR6, IGHG2, RP11-291B21.2, TC2N, PRDX4, BCO2, HSPA6, TPSB2, CD28, ZFY, AIM1, TCL1A, IGHV1-2, TRABD2A, DMBT1, IGLV1-51, RP5-1028K7.2, IGLV2-11, TSHZ2, IGHG4, UTY, POU2AF1, CST7, ZNF10, AC005480.1, RNF19A, CCL20, CCL11, RNASE6, ZNF90, RP5-887A10.1, SLC16A1-AS1, DCN, ZSWIM4, KLHL6-AS1, ITM2C, USP9Y, LINC00402, GIMAP7, NTS, CREM, MIR4435-2HG, UBC, IGHG3, SEPP1, RGS1, VPREB3, ICOS, FAM65B, 1-Mar.1, TTTY15, JCHAIN, MT1E, AC008079.10, DNAJB1, RGCC, TRBC2, IGFBP7, TRBC1, HOPX, DUSP2, COL1A2, TRDC, PLAC8, CC2D2A, RP11-18H21.1, CST3, SNHG25, IGLC6, ODF2L, PGAP1, PHLDA1, ADAMDEC1, DUSP4, CH507-42P11.8, IGHGP, IGLC2, AC129778.2, ID3, IGLC5, IL17A, CALD1, IGKV3-15, GZMK, IGHA2, FAM118A, MARCKSL1, THADA, PRAP1, RP11-3P17.5, TXNIP, FYN, GZMH, TNFRSF18, LINC00926, CYBA, ZFX, MZB1, KDM5D, AC005540.3, MT1X, MTRNR2L8, JUNB, AIF1, MTRNR2L11, RP11-693J15.5, IGLV1-41, DUSP1, IL32, LAPTM5, NEAT1, SPARCL1, IL1B, HERPUD1, TRAT1
Scatter plot of selected markers
[28]:
sc.settings.set_figure_params(dpi=50, facecolor='white')
sc.pl.umap(adata, color=selected_markers[0:40], frameon=False, ncols=5, color_map="inferno")

[29]:
sc.pl.umap(adata, color=selected_markers[40:80], frameon=False, ncols=5, color_map="inferno")

[30]:
sc.pl.umap(adata, color=selected_markers[80:120], frameon=False, ncols=5, color_map="inferno")

[31]:
sc.pl.umap(adata, color=selected_markers[120:160], frameon=False, ncols=5, color_map="inferno")

[32]:
sc.pl.umap(adata, color=selected_markers[160:200], frameon=False, ncols=5, color_map="inferno")

[33]:
sc.pl.umap(adata, color=selected_markers[200:240], frameon=False, ncols=5, color_map="inferno")

[34]:
sc.pl.umap(adata, color=selected_markers[240:250], frameon=False, ncols=5, color_map="inferno")
Violin plot of some markers
[35]:
major_cell_type_dict = {
'IgA plasma cells': 'Plasma',
'Tregs': "T",
'Mast cells': "Mast",
'TRM': "T",
'Group3 ILC': 'ILC3',
'B cells': "B",
'DC2': 'Phagocytes',
'T (gd)': "T",
'IgM plasma cells': 'Plasma',
'TFH-like T cells': "T",
'Group1 ILC': 'ILC1',
'Fibroblasts': "Stromal",
'Macrophages': 'Phagocytes',
'IgG plasma cells': 'Plasma',
'DC2 CD206+': 'Phagocytes',
'DC1': 'Phagocytes',
'ACKR1+ endothelial cells': "Stromal",
'Central Memory T cells': "T",
'Naive T cells': "T",
'Cytotoxic T cells': "T",
'Highly activated T cells': "T",
'Pericytes': "Stromal",
'Plasmablasts': 'Plasma',
'CD36+ endothelial cells': "Stromal",
'Lymphatics': "Stromal",
'Smooth muscle cells': "Stromal",
'Glial Cells': "Stromal"
}
[36]:
adata.obs['major cell types'] = adata.obs['Primary Lineages'].apply(major_cell_type_dict.__getitem__)
[37]:
sc.settings.set_figure_params(dpi=80, facecolor='white')
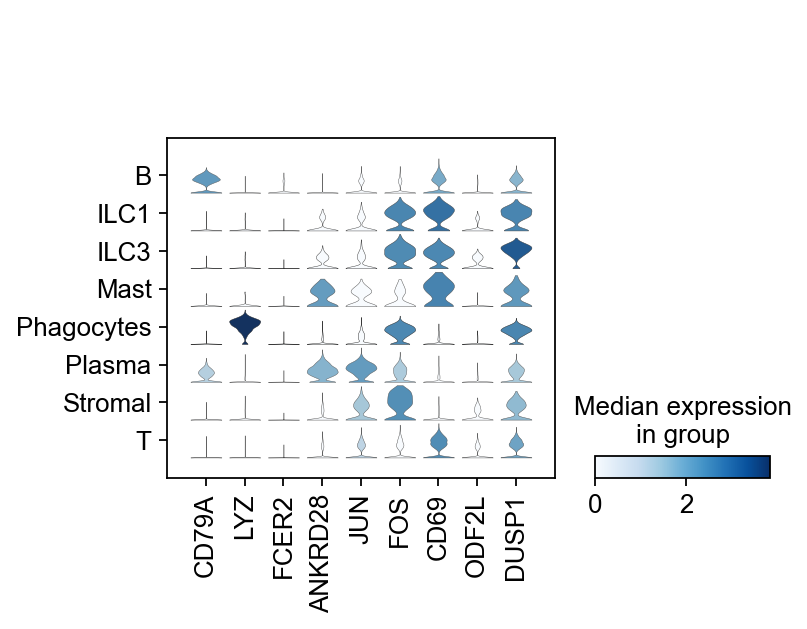
sc.pl.stacked_violin(adata, ['CD79A', 'LYZ', 'FCER2', 'ANKRD28', 'JUN', 'FOS', 'CD69', 'ODF2L', 'DUSP1'], groupby="major cell types")
... storing 'major cell types' as categorical
[38]:
sc.logging.print_versions()
WARNING: If you miss a compact list, please try `print_header`!
-----
anndata 0.7.4
scanpy 1.6.0
sinfo 0.3.1
-----
PIL 7.2.0
anndata 0.7.4
backcall 0.2.0
cairo 1.19.1
cffi 1.14.2
colorama 0.4.3
cycler 0.10.0
cython_runtime NA
dateutil 2.8.1
decorator 4.4.2
get_version 2.1
h5py 2.10.0
igraph 0.8.2
importlib_metadata 1.7.0
ipykernel 5.3.4
ipython_genutils 0.2.0
ipywidgets 7.5.1
jedi 0.17.2
jinja2 2.11.2
joblib 0.16.0
kiwisolver 1.2.0
legacy_api_wrap 1.2
leidenalg 0.8.1
llvmlite 0.33.0+1.g022ab0f
markupsafe 1.1.1
matplotlib 3.3.1
mkl 2.3.0
mpl_toolkits NA
natsort 7.0.1
nt NA
ntsecuritycon NA
numba 0.50.1
numexpr 2.7.1
numpy 1.19.1
packaging 20.4
pandas 1.1.1
parso 0.7.0
pickleshare 0.7.5
pkg_resources NA
prompt_toolkit 3.0.7
pycparser 2.20
pygments 2.6.1
pyparsing 2.4.7
pythoncom NA
pytz 2020.1
pywintypes NA
scanpy 1.6.0
scipy 1.5.2
scmer NA
seaborn 0.11.0
setuptools_scm NA
sinfo 0.3.1
six 1.15.0
sklearn 0.23.2
sphinxcontrib NA
statsmodels 0.11.1
storemagic NA
tables 3.6.1
texttable 1.6.2
torch 1.6.0
tornado 6.0.4
tqdm 4.48.2
traitlets 4.3.3
umap 0.4.6
wcwidth 0.2.5
win32api NA
win32com NA
win32security NA
zipp NA
zmq 19.0.1
-----
IPython 7.18.1
jupyter_client 6.1.6
jupyter_core 4.6.3
notebook 6.1.1
-----
Python 3.7.7 (default, May 6 2020, 11:45:54) [MSC v.1916 64 bit (AMD64)]
Windows-10-10.0.17763-SP0
12 logical CPU cores, Intel64 Family 6 Model 158 Stepping 10, GenuineIntel
-----
Session information updated at 2020-11-11 23:49
[ ]: